・Basic
Research・・Current Issue・ ・Achieve・ ・Search
Articles・ ・Online
Submission・ ・About IJO・
Use of high-throughput targeted exome sequencing
in genetic diagnosis of Chinese family with congenital cataract
Ming-Fu
Ma1, Lian-Bing Li1, Yun-Qi Pei2, Zhi Cheng2
1Key Laboratory of Birth Defects and Reproductive
Health of the National Health and Family Planning Commission (Chongqing
Population and Family Planning Science and Technology Research Institute),
Chongqing 400020, China
2Department of Cell Biology and Genetics, Chongqing
Medical University, Chongqing 400016, China
Co-first
authors: Ming-Fu Ma and
Lian-Bing Li
Correspondence to: Zhi Cheng. Department of Cell Biology and Genetics, Chongqing
Medical University, Chongqing 400016, China. zcheng@cqmu.edu.cn
Received: 2015-06-03
Accepted: 2015-07-28
AIM: To identify disease-causing mutation in a
congenital cataract family using enrichment
of targeted genes combined with next-generation sequencing.
METHODS: A total of 371 known genes related to inherited eye diseases of the
proband was selected and captured, followed by high-throughput sequencing. The
sequencing data were analyzed by established bioinformatics pipeline.
Validation was performed by Sanger sequencing.
RESULTS: A recurrent heterozygous non-synonymous mutation c.130G>A (p.V44M)
in the GJA3 gene was identified in
the proband. The result was confirmed by Sanger sequencing. The mutation showed
co-segregation with the disease phenotype in the family but was not detected in
unaffected controls.
CONCLUSION: Targeted exome sequencing is a rapid, high-throughput and
cost-efficient method for screening known genes and could be applied to the
routine gene diagnosis of congenital cataract.
KEYWORDS: genetic diagnosis; targeted exome
sequencing; congenital cataract
DOI:10.18240/ijo.2016.05.02
Citation: Ma MF, Li LB, Pei YQ, Cheng Z. Use of
high-throughput targeted exome sequencing in genetic diagnosis of Chinese
family with congenital cataract. Int
J Ophthalmol 2016;9(5):650-654
Congenital
cataract is the leading cause of visual impairment and blindness in children.
It has an estimated incidence of 1-6 per 10 000 live births and accounts for
nearly 10% of irreversible childhood blindness worldwide[1-2]. For affected children, early diagnosis is
important because timely and appropriate intervention can obtain good visual
function[3].
Approximately
one-third of congenital cataract cases are believed to be hereditary, and most
occur in an autosomal dominant pattern[4].
To date, more than 40 loci have been mapped in congenital cataracts and 32
genes have been identified[5].
These genes contain a total of 250 coding fragments (Table 1). Therefore,
genetic diagnosis through traditional approaches, such as direct sequencing is
time-consuming and costly. A more efficient method to detect the genetic
defects is needed.
Table 1
Known causative genes of congenital cataract
|
No. |
Locus |
Gene |
Coding exons |
|
1 |
1p32 |
1FOXE3 |
1 |
|
2 |
1p36 |
EPHA2 |
18 |
|
3 |
1q21.1 |
GJA8 |
2 |
|
4 |
2q33.3 |
CRYGC |
3 |
|
5 |
2q33.3 |
CRYGD |
3 |
|
6 |
3p21.31 |
FYCO1 |
22 |
|
7 |
3q22.1 |
BFSP2 |
7 |
|
8 |
3q27.3 |
1CRYGS |
3 |
|
9 |
6p24.2 |
GCNT2 |
11 |
|
10 |
7q34 |
AGK |
17 |
|
11 |
8q13.3 |
EYA1 |
20 |
|
12 |
9q22.33 |
TDRD7 |
17 |
|
13 |
10p13 |
1VIM |
9 |
|
14 |
10q24.32 |
PITX3 |
4 |
|
15 |
11q25 |
JAM3 |
9 |
|
16 |
11q22.3-q23.1 |
CRYAB |
3 |
|
17 |
12q13 |
MIP |
4 |
|
18 |
13q12.11 |
GJA3 |
3 |
|
19 |
16p13.2 |
1TMEM114 |
4 |
|
20 |
16q21 |
HSF4 |
15 |
|
21 |
16q22-q23 |
MAF |
2 |
|
22 |
17q11.2 |
CRYBA1 |
6 |
|
23 |
19q13.33 |
FTL |
4 |
|
24 |
19q13.4 |
LIM2 |
5 |
|
25 |
20p12.1 |
BFSP1 |
13 |
|
26 |
20q11.22 |
CHMP4B |
5 |
|
27 |
21q22.3 |
CRYAA |
4 |
|
28 |
22q11.23 |
CRYBB2 |
7 |
|
29 |
22q11.23 |
CRYBB3 |
6 |
|
30 |
22q12.1 |
CRYBB1 |
6 |
|
31 |
22q12.1 |
CRYBA4 |
7 |
|
32 |
Xp22.13 |
NHS |
10 |
|
Total |
250 |
1Genes
which were not captured in the current study.
With the
progresses on next-generation sequencing and bioinformatics, whole exome
sequencing has been proved to be a powerful tool for the genetic diagnosis of
both Mendelian and complex diseases[6-7].
And it has been successfully applied in identifying disease-causing genes and
mutations for congenital cataract[8-9].
However, the large amount of information, subsequent difficult data processing
and high cost limit its potential application in clinical practice. In this
study, we utilized high-throughput targeted exome sequencing (TES) to study
genetic defects in a congenital cataract family and attempt to establish a
strategy feasible to genetic diagnosis of congenital cataract patients.
Subjects
The proband, a
7-year-old boy, was diagnosed with bilateral cataracts at Affiliated Hospital
of Chongqing Population and Family Planning Science and Technology Research
Institute. A family history revealed six members in three generations. Five
family members participated in the study. The research protocol was approved by
the ethics committee of Chongqing Population and Family Planning Science and
Technology Research Institute. All participants from the family provided their
written consent for participation in the research. And they didn't receive any
stipend. The study was conducted according to the principles in the Declaration
of Helsinki. Peripheral blood samples were collected from all study
participants.
Targeted
Capture Preparation and Next-generation Sequencing Genomic DNA was extracted from whole blood using
TIANamp Blood DNA Kit (Tiangen Biotech Co. Ltd., Beijing, China). The DNA was
quantified with Nanodrop 2000 (Thermo Fisher Scientific, MA, USA). A minimum of
3 μg DNA was used for the indexed Illumina libraries according to manufacturer’s
protocol. The final library size 350-450 bp including adapter sequences was
selected. The coding exons and flanking regions of 371 genes related to
inherited eye diseases were selected and captured using a GenCap custom
enrichment kit[10]. The methods used for DNA target capture,
enrichment and elution followed previously described protocols [11-13]. The eluted DNA was finally amplified as follows: 98℃ for 30s; 98℃ for 25s, 65℃ for 30s,
72℃ for 30s (15 cycles); 72℃ for 5min. The polymerase chain reaction (PCR) product was purified using SPRI beads (Beckman Coulter,
CA, USA) according to manufacturer’s protocol. The purified product was
sequenced on Illumina Solexa HiSeq 2000 sequencer (Illumina, CA, USA).
Bioinformatics
Analysis After sequencing, the Solexa QA package and
the cutadapt program (http://code.google.com/p/cutadapt/) were used to
filtering out the low quality reads and adaptor sequences[14]. The SOAPaligner program was used to align the clean
read sequences to the human reference genome (hg19)[15].
The PCR duplicates were removed by the
Picard software and single nucleotide polymorphisms (SNPs) were identified
using the SOAPsnp program[15-16]. Subsequently, reads were realigned to the
reference genome using the Burrows-Wheeler alignment program, and insertions or
deletions (InDels) were identified with the Genome Analysis Toolkit[17-18]. The identified SNPs and InDels were annotated using
the Exome-assistant program (http://122.228.158.106/exomeassistant).
MagicViewer was used to view the short read alignment and validate the
candidate SNPs and InDels[19]. Finally, nonsynonymous variants were
evaluated by four algorithms, SIFT, PolyPhen_2, MutationTaster and GERP++, to
determine pathogenicity.
Expanded
Validation Sanger sequencing was used to confirm the
potential pathogenic variants detected by TES. Primers were used to amplify
coding exons containing the candidate variants and their flanking regions
(GJA3-F: 5’-CGGTGTTCATGAGCATTTTC-3’ and GJA3-R: 5’-CTCTTCAGCTGCTCCTCCTC-3’).
The PCR products were sequenced with the ABI3730 Automated Sequencer (Applied
Biosystems, CA, USA). DNA samples from all participating members of the family
were analyzed. The sequencing results were analyzed using Chromas software and
compared with the reference sequences in the NCBI database.
Clinical
Evaluation Five family members of a three-generation
Chinese family with a history of cataracts participated in the study (three
affected and two unaffected individuals; Figure 1). All patients in this family
had bilateral cataracts. The proband, a 7-year-old boy,had been diagnosed with bilateral cataracts at the age
of 6mo. His grandfather (I:1) and mother (II:1) also had poor vision in their
childhood. The boy’s best corrected visual acuity was 0.2/0.4. There was no
family history of other ocular or systemic abnormalities. To date, all of the
affected individuals have had cataract surgery.
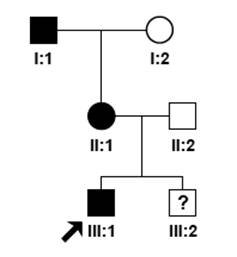
Figure 1 Pedigree
of family with congenital cataract Circles represent
females, while squares indicate males. Affected individuals are denoted by
black symbols. The arrow points to the proband. The phenotypes of proband’s
brother is unknown while the study was carried on.
Targeted Exome Sequencing and Co-segregation Analysis
Identified Causative Mutation The genomic DNA of the proband was
subjected to TES. Three hundred and seventy-one genes related to inherited eye
diseases were analyzed including 28 genes involved in congenital cataract. The
average sequencing depth on the targeted regions was 215.45. And the sample had
95.30% coverage of the targeted regions. Meanwhile, 89.40% and 81.50% targeted
exons are covered with at least 4 and 10 reads, respectively (Table 2). A total
of 23 variants were identified in the 28 known cataract genes. After excluding
variants reported in HapMap 28 and the SNPs release of the 1000 Genome Project
with minor allele frequency >0.05, a previously reported heterozygous
non-synonymous mutation c.130G>A (p.V44M) was discovered in exon 2 of GJA3 (Figure 2). It was predicted to be
damaging by bioinformatic software tools. The mutation was further confirmed in
the proband by Sanger sequencing. It was also found in two other affected
family members but was absent in unaffected family members (Figure 3). The
mutation was not detected in the 100 unrelated controls either[20]. So, it co-segregated with the congenital cataract
phenotype. Additionally, a heterozygous non-synonymous mutation c.605A>G
(p.E202G) in GCNT2 was also detected
in the proband.
Table 2 Data summary of the
targeted exome sequencing
|
Sample |
Proband |
|
Raw data (Mb) |
418.5 |
|
Clean data (Mb) |
415.78 |
|
Aligned |
99.8 |
|
Initial bases on target |
951791 |
|
Base covered on target |
907495 |
|
Coverage of target region |
95.30% |
|
Total effective yield (Mb) |
332.63 |
|
Effective sequence on target (Mb) |
205.06 |
|
Fraction of effective bases on target |
61.60% |
|
Average sequencing depth on target |
215.45 |
|
Fraction of target covered with at least 4× |
89.40% |
|
Fraction of target covered with at least 10× |
81.50% |
|
Fraction of target covered with at least 20× |
72.90% |
|
Duplication rate (%) |
17.4741 |
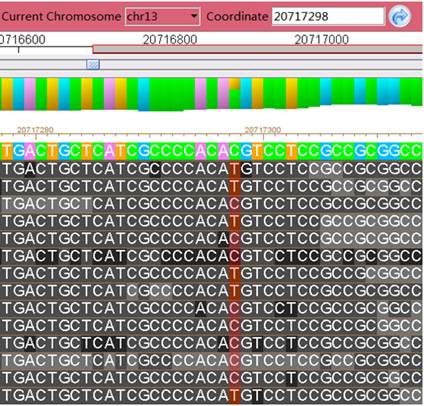
Figure 2 GJA3
mutation identified by TES in the proband.
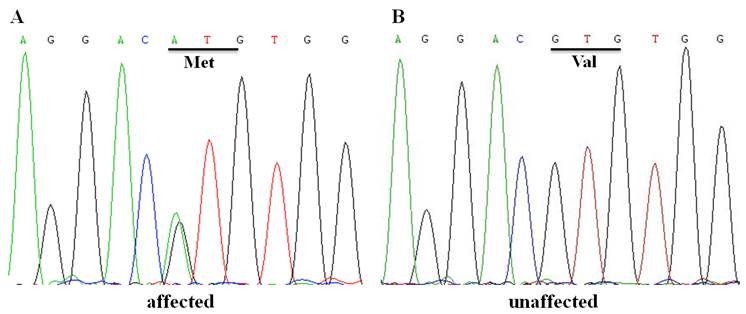
Figure 3 DNA sequences of GJA3 in affected and unaffected
individuals A: The DNA sequence
chromatograms showing the heterozygous c.130G>A transition that replaces
valine by methionine at codon 44 in an affected individual; B: The DNA sequence
chromatograms of an unaffected individual.
Congenital cataract is clinically and
genetically highly heterogeneous. It is known that different mutations in the
same gene can cause similar phenotypes, while the same mutation in a single
gene can lead to different cataract patterns within the families[21]. Together with the fact that a huge number of coding
exons exist in known genes, traditional screening of each region is impractical
for genetic diagnosis and testing in clinical practice. In this study, we used
enrichment of targeted genes in combination with high-throughput sequencing to
screen mutations in a Chinese pedigree with congenital cataract. And we
successfully identified a previously reported recurrent disease-causing mutation.
The results were validated by Sanger sequencing. It demonstrated the robustness
and potential application of this approach in clinical genetic diagnosis of
congenital cataract.
At present, whole exome sequencing has
emerged as a useful new method in genetic diagnosis of congenital cataract[8-9]. Although its cost is now falling, it’s still too high
for clinical practice. In the current study, our approach only captured and
analyzed the exons of targeted genes. The cost was saved at least 50% compared
with whole exome sequencing. And a large amount of work in data analysis was
saved simultaneously. Furthermore, the sequencing depth was increased as a
result of the decreased targeted region. Therefore, our approach is more
feasible than whole exome sequencing in clinical practice.
GJA3, which encodes Connexin 46, is mainly
expressed in lens fiber cells and plays a key role in maintaining normal lens
transparency. Knock-out of GJA3 in
mice leads to different degree of cataracts depending on genetic background[22-23]. To date, more than 20 mutations of GJA3 have been reported to cause
congenital cataract[24]. Utilizing TES, we identified a
heterozygous mutation of GJA3 (p.V44M)
in a Chinese family. The same mutation had been reported in a Han Chinese
family and in a Caucasian American family[20,25]. It
indicated that the V44 might be a mutation hotspot.
Through TES, a heterozygous mutation of
GCNT2 (p.E202G) was also detected in
the proband. But it could not be the causative mutation of the family because
the GCNT2 had been demonstrated to
associate with autosomal recessive cataract [26-27]. It
indicated that TES could provide complete information of genetic defects which
might be undetectable by traditional methods.
In summary, we diagnosed congenital cataract
genetically using enrichment of targeted genes combined with next-generation
sequencing and proved that it is a rapid, high-throughput and cost-efficient
method. This strategy can be also used to genetically diagnose of other
monogenic diseases.
ACKNOWLEDGEMENTS
Foundations:
Supported by the National
Natural Science Foundation of China (No.81300463; No.81130051); Fundamental
Research Funds for Non-profit Public Scientific Research Institutions of
Chongqing (No.2012CSTC-jbby-01704); Natural Science Foundation Project of CQ
CSTC (No.cstc2013jcyjA10086); Promotion Program for Young and Middle-aged
Teacher in Scientific Research of Basic Medicine College, Chongqing Medical
University (No.JC201306).
Conflicts
of Interest: Ma MF, None; Li LB, None; Pei YQ,
None; Cheng Z, None.
1 Francis
PJ, Berry V, Bhattacharya SS, Moore AT. The genetics of childhood cataract. J Med Genet 2000;37(7):481-488. [CrossRef]
2 Apple
DJ, Ram J, Foster A, Peng Q. Elimination of cataract blindness: a global
perspective entering the new millenium. Surv
Ophthalmol 2000;45(Suppl 1):S1-196. [PubMed]
3 Chan WH,
Biswas S, Ashworth JL, Lloyd IC. Congenital and infantile cataract: aetiology
and management. Eur J Pediatr 2012;171(4):625-630. [CrossRef] [PubMed]
4 Reddy
MA, Francis PJ, Berry V, Bhattacharya SS, Moore AT. Molecular genetic basis of
inherited cataract and associated phenotypes. Surv Ophthalmol 2004;49(3):300-315. [CrossRef] [PubMed]
5 Shiels
A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map. Mol Vis 2010;16:2007-2015. [PMC free article] [PubMed]
6 Bilgüvar
K, Oztürk AK, Louvi A, et al.
Whole-exome sequencing identifies recessive WDR62 mutations in severe brain
malformations. Nature
2010;467(7312):207-210. [CrossRef] [PubMed] [PMC free article]
7 Pugh TJ,
Weeraratne SD, Archer TC, et al.
Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012;488(7409):106-110. [CrossRef] [PubMed] [PMC free article]
8 Kondo Y,
Saitsu H, Miyamoto T, Lee BJ, Nishiyama K, Nakashima M, Tsurusaki Y, Doi H,
Miyake N, Kim JH, Yu YS, Matsumoto N. Pathogenic mutations in two families with
congenital cataract identified with whole-exome sequencing. Mol Vis 2013;19:384-389. [PMC free article] [PubMed]
9 Reis LM,
Tyler RC, Muheisen S, Raggio V, Salviati L, Han DP, Costakos D, Yonath H, Hall
S, Power P, Semina EV. Whole exome sequencing in dominant cataract identifies a
new causative factor, CRYBA2, and a variety of novel alleles in known genes. Hum Genet 2013;132(7):761-770. [CrossRef] [PubMed] [PMC free article]
10 Yang L,
Wu L, Yin X, Chen N, Li G, Ma Z. Novel mutations of CRB1 in Chinese families
presenting with retinal dystrophies. Mol
Vis 2014;20:359-367. [PMC free article] [PubMed]
11 He J,
Wu J, Jiao Y, Wagner-Johnston N, Ambinder RF, Diaz LA Jr, Kinzler KW,
Vogelstein B, Papadopoulos N. IgH gene rearrangements as plasma biomarkers in
Non- Hodgkin's lymphoma patients. Oncotarget
2011;2(3):178-185. [CrossRef] [PubMed] [PMC free article]
12 Wu J,
Matthaei H, Maitra A, Dal Molin M, Wood LD, Eshleman JR, Goggins M, Canto MI,
Schulick RD, Edil BH, Wolfgang CL, Klein AP, Diaz LA Jr, Allen PJ,Schmidt CM,
Kinzler KW, Papadopoulos N, Hruban RH, Vogelstein B. Recurrent GNAS mutations
define an unexpected pathway for pancreatic cyst development. Sci Transl Med 2011;3(92):92ra66. [CrossRef]
13 Huang
XF, Xiang P, Chen J, Xing DJ, Huang N, Min Q, Gu F, Tong Y, Pang CP, Qu J, Jin
ZB. Targeted exome sequencing identified novel USH2A mutations in Usher
syndrome families. PLoS One
2013;8(5):e63832. [CrossRef] [PubMed] [PMC free article]
14 Cox MP,
Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina
second-generation sequencing data. BMC
Bioinformatics 2010;11:485. [CrossRef] [PubMed] [PMC free article]
15 Li R,
Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast
tool for short read alignment. Bioinformatics
2009;25(15):1966-1967. [CrossRef] [PubMed]
16 Li H,
Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin
R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map
format and SAMtools. Bioinformatics
2009;25(16):2078-2079. [CrossRef] [PubMed] [PMC free article]
17 Li H,
Durbin R. Fast and accurate short read alignment with Burrows-Wheeler
transform. Bioinformatics
2009;25(14):1754-1760. [CrossRef] [PubMed] [PMC free article]
18
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis
AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM,
Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for
variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43(5):491-498. [CrossRef]
[PubMed] [PMC free article]
19 Hou H, Zhao F, Zhou L, Zhu E, Teng H, Li X, Bao Q, Wu J, Sun Z.
MagicViewer: integrated solution for next-generation sequencing data
visualization and genetic variation detection and annotation. Nucleic Acids Res 2010;38(Web Server
issue):W732-736.
20 Zhou Z,
Hu S, Wang B, Zhou N, Zhou S, Ma X, Qi Y. Mutation analysis of congenital
cataract in a Chinese family identified a novel missense mutation in the
connexin 46 gene (GJA3). Mol Vis
2010;16:713-719. [PMC free article] [PubMed]
21 Santana
A, Waiswo M. The genetic and molecular basis of congenital cataract. Arq Bras Oftalmol 2011;74(2):136-142. [CrossRef] [PubMed]
22 Gong X,
Li E, Klier G, Huang Q, Wu Y, Lei H, Kumar NM, Horwitz J, Gilula NB. Disruption
of alpha3 connexin gene leads to proteolysis and cataractogenesis in mice. Cell 1997;91(6):833-843. [CrossRef]
23 Gong X,
Agopian K, Kumar NM, Gilula NB. Genetic factors influence cataract formation in
alpha 3 connexin knockout mice. Dev Genet
1999;24(1-2):27-32. [CrossRef]
24 Guo Y,
Yuan L, Yi J, Xiao J, Xu H, Lv H, Xiong W, Zheng W, Guan L, Zhang J, Xiang H,
Qi Y, Deng H. Identification of a GJA3 mutation in a Chinese family with
congenital nuclear cataract using exome sequencing. Indian J Biochem Biophys 2013;50(4):253-258. [PubMed]
25 Bennett
TM, Shiels A. A recurrent missense mutation in GJA3 associated with autosomal
dominant cataract linked to chromosome 13q. Mol
Vis 2011;17:2255-2262. [PMC free article] [PubMed]
26 Yu LC,
Twu YC, Chou ML, Reid ME, Gray AR, Moulds JM, Chang CY, Lin M. The molecular
genetics of the human I locus and molecular background explain the partial
association of the adult i phenotype with congenital cataracts. Blood 2003;101(6):2081-2088. [CrossRef] [PubMed]
27 Pras E,
Raz J, Yahalom V, Frydman M, Garzozi HJ, Pras E, Hejtmancik JF. A nonsense
mutation in the glucosaminyl (N-acetyl) transferase 2 gene (GCNT2): association
with autosomal recessive congenital cataracts. Invest Ophthalmol Vis Sci 2004;45(6):1940-1945.<bb> [CrossRef]
[Top]