íĄBasic ResearchíĄíĄCurrent IssueíĄ íĄAchieveíĄ íĄSearch ArticlesíĄ íĄOnline SubmissioníĄ íĄAbout IJOíĄ
In silico
analysis of a disease-causing mutation in PCDH15 gene in a
consanguineous Pakistani family with Usher phenotype
Shamim Saleha1, Muhammad Ajmal2,
Muhammad Jamil1, Muhammad Nasir2, Abdul Hameed2
1Department of Biotechnology and Genetic Engineering, Kohat
University of Science and Technology, Kohat 26000, Khyber Pakhtunkhwa, Pakistan
2Institute of Biomedical and Genetic Engineering (IBGE),
Islamabad 44000, Pakistan
Correspondence to: Shamim Saleha. Department of
Biotechnology and Genetic Engineering, Kohat University of Science and
Technology, Kohat 26000, Khyber Pakhtunkhwa, Pakistan. shamimsaleha@yahoo.com
Received: 2015-04-24
Accepted: 2015-08-06
Abstract
AIM: To map Usher phenotype in
a consanguineous Pakistani family and identify disease-associated mutation in a
causative gene to establish phenotype-genotype correlation.
METHODS: A consanguineous
Pakistani family in which Usher phenotype was segregating as an autosomal
recessive trait was ascertained. On the basis of results of clinical
investigations of affected members of this family disease was diagnosed as
Usher syndrome (USH). To identify the locus responsible for the Usher phenotype
in this family, genomic DNA from blood sample of each individual was genotyped
using microsatellite Short
Tandem Repeat (STR) markers for the known Usher syndrome loci. Then
direct sequencing was performed to find out disease associated mutations in the
candidate gene.
RESULTS: By genetic linkage
analysis, the USH phenotype of this family was mapped to PCDH15 locus on
chromosome 10q21.1. Three different point mutations in exon 11 of PCDH15
were identified and one of them, c.1304A>C was found to be segregating with
the disease phenotype in Pakistani family with Usher phenotype. This,
c.1304A>C transversion mutation predicts an amino-acid substitution of
aspartic acid with an alanine at residue number 435 (p.D435A) of its protein
product. Moreover, in silico analysis revealed conservation of aspartic acid at
position 435 and predicated this change as pathogenic.
CONCLUSION:
The
identification of c.1304A>C pathogenic mutation in PCDH15 gene and
its association with Usher syndrome in a consanguineous Pakistani family is the
first example of a missense mutation of PCDH15 causing USH1 phenotype.
In previous reports, it was hypothesized that severe mutations such as
truncated protein of PCDH15 led to the Usher I phenotype and that
missense variants are mainly responsible for non-syndromic hearing impairment.
KEYWORDS: deafness and blindness;
Usher syndrome; causative gene; missense mutation; Pakistani family
DOI:10.18240/ijo.2016.05.04
Citation: Saleha S, Ajmal M, Jamil M, Nasir M, Hameed A. In
silico analysis of a disease-causing mutation in PCDH15 gene in a
consanguineous Pakistani family with Usher phenotype. Int J Ophthalmol 2016;9(5):662-668
INTRODUCTION
Usher syndrome (USH) is inherited as an autosomal recessive
trait and is characterized by a loss of vision due to retinitis pigmentosa (RP)
and bilateral sensorineural deafness[1-3]. USH
represents the most common genetic cause of deafness and blindness among
children. Affected children are born deaf and
progressively develop pigmentary retinopathy leading to blindness[4].
USH is classified into three clinical subtypes, designated as
types I, type II, and type III. These types are distinguished by their severity
and the age of onset of the disease[5]. USH type I (USH1) is the most
severe and is characterized by severe to profound congenital hearing
impairment, vestibular dysfunction, and pre-pubertal onset of RP; type II
(USH2) is the most frequent form and is characterized by moderate to severe
hearing impairment, normal vestibular function, and teenage onset of RP. USH type III (USH3) presents
with progressive hearing loss and variable onset of RP and vestibular function[6].
USH1 is an autosomal recessive disorder
and its genetic heterogeneity is well established as different mutant genes,
causing USH, have been identified. To date, seven different loci USH1B (11q13.5), USH1C (11p15.1), USH1D (10q21-q22), USH1E (21q21), USH1F (10q21-q22), USH1G (17q24-q25), and USH1H (15q22-q23) have been
reported to cause USH1. Genes at five of these loci, MYO7A (USH1B), USH1C (USH1C), CDH23 (USH1D), PCDH15 (USH1F), and USH1G
(USH1G) have been identified[7]. Mutations of four USH1 genes MYO7A,
USH1C, CDH23, and PCDH15 are also reported to cause
nonsydromic deafness, DFNB2, DFNB18, DFNB12 and DFNB23, respectively[8-9].
In this study, the USH1F locus on chromosome 10q21.1 was
mapped by genetics linkage analysis in a consanguineous family with Usher
phenotype from Khyber Pakhtunkhwa, Pakistan. This region of chromosome 10
harbors PCDH15 gene. It was reported previously that protein truncating
mutations of PCDH15 cause USH1F and missense mutation of PCDH15
were associated with isolated deafness (DFNB23) only[10]. On
sequencing of PCDH15 gene, we identified three sequence variants in exon
11 of PCDH15 gene. One of the sequence variant, c.1304A>C was
observed to be segregating with the disease trait and was not present in
ethnically matched controls. This pathogenic c.1304A>C mutation predicts the
substitution of an amino acid residue aspartic acid to alanine at codon 435
(p.435D>A). The association of a substitution mutation of PCDH15 to
USH trait in a Pakistani family is the first report of an association of
substitution mutation with the USH trait.
MATERIALS AND METHODS
Sample Collection and Genomic DNA Isolation This study was approved by
Advance Studies and Research Board, Kohat University of Science and Technology,
Khyber Pakhtunkhwa, Pakistan and Institute of Biomedical and Genetic
Engineering, Islamabad, Pakistan. We studied an autosomal recessive
consanguineous three generation family with USH. This family was collected from
the Khyber Pakhtunkhwa province of Pakistan, where cousin marriages are
commonly practiced. On the basis of clinical history and the results of
ophthalmologic, audiometric, and vestibular tests, disease was diagnosed as
USH. The clinical information of the affected individuals is presented in Table
1. Blood samples from affected individuals, their parents and clinically normal
siblings of the family members were collected with informed consent. Blood
samples were also collected from 100 ethnically matched unrelated normal
Pakistani individuals and were used as controls for allele frequencies and
confirmation of disease associated mutation. Genomic DNA was extracted from
peripheral blood by the standard phenol chloroform extraction procedure [11].
Table 1 A summary of
clinical findings of affected individuals with USH
|
Pedigree code |
Age (a) |
Clinical findings |
||
|
Hearing loss |
Vestibular balance |
Blindness |
||
|
USHR506 |
15 |
Congenital, bilateral and profound |
Disturb |
Profound |
|
USHR507 |
13 |
Congenital, bilateral and profound |
Disturb |
Profound |
|
USHR508 |
11 |
Congenital, bilateral and profound |
Disturb |
Progressive |
|
USHR512 |
10 |
Congenital, bilateral and profound |
Disturb |
Progressive |
Microsatellite and Linkage Analysis To identify the locus responsible for the disease
in this family, genomic DNA from each individual was genotyped using
microsatellite Short Tandem Repeat (STR) markers for the known USH loci (Table 2). Each STR
marker was amplified by polymerase chain reaction (PCR). PCR reactions were
performed in a 10 mL volume, each containing 1.5 mmol/L MgCl2, 0.6 mmol/L of each primer, 0.2 mmol/L dNTPs, 1 U Taq DNA polymerase
and PCR buffer [16 mmol/L (NH4) 2SO4, 67 mmol/LTris-HCI (pH 8.8),
and 0.01% of the nonionic detergent Tween-20] (Bio-line, London, UK).
Amplification was performed with an initial denaturation for 4min at 94íŠ, followed by 35 cycles of
denaturation at 94íŠ for 45s, annealing at 55íŠ for 45s, extension at 72íŠ for 45s and a final extension
at 72íŠ for 5min. The PCR products were separated on 10%
non-denaturing polyacrylamide gels (Protogel; National Diagnostics, Edinburgh,
Scotland, UK). The gel was stained with ethidium bromide and photographed under
ultraviolet (UV) illumination. Alleles were
assigned to individuals and genotypic data was used to calculate the LOD scores
using the Cyrillic and MLINK software programme (Version 5.2,
ftp://linkage.rockefeller.edu/software/linkage/).
Table 2 Known USH loci and genetic
markers used in this study
|
Locus I.D. |
Chromosomal region |
Gene |
STR markers used for exclusion studies |
|
USH2A |
1q41 |
Usherin |
D1S2141,
D1S549 |
|
USH1F |
10q21.1 |
PCDH15 |
D10S1220,
D10S1225, D10S1221, D10S1208, GATA121A08 |
|
USH1C |
11q15.1 |
Harmonin |
D11S1981,
ATA34E08 |
|
USH1B |
11q13.5 |
Myosin
1B |
D11S2371,
D11S2002, D11S2000 |
|
USH1G |
17q24-q25 |
Scaffold
protein |
ATA43A10,
D17S784, D17S949 |
Polymerase
Chain Reaction Amplification of Genomic DNA and Mutation Screening by Direct
DNA Sequencing PCR amplification of the PCDH15 gene was performed with primers
spanning all 35 exons[12-13]. PCR amplification was
performed in a 50 µL reaction volume containing 250 ng of genomic DNA,
amplification buffer containing 600 nmol/L of each primer, 1.5 mmol/L MgCl2, 200 mmol/L of dNTPs and 2.5 U Taq
polymerase (Applied Biosystems, Warrington, UK) in an PxE thermal cycler
(Hybaid, Basingstoke, UK). The amplification conditions were 95íŠ for 5min, followed by 35
cycles of 95íŠ for
45s, primer specific annealing temperature (55íŠ-65íŠ) for 45s, 72íŠ for 45s. Aliquots (5 µL) of the PCR products were analyzed by 2.5%
agarose gel electrophoresis. PCR products were then purified using GeneJetTM PCR
purification kit (Fermentas Life Sciences, Hanover, MD, USA) and sequenced directly using Big Dye® Terminator v3.1 cycle
sequencing kit in an ABI 3130 genetic analyzer (Applied Biosystems, Foster
City, CA, USA). Potential mutations were confirmed by bi-directional sequencing
and assessing 100 control samples having ethnic backgrounds matching to
patients.
In silico Analysis of the PCDH15 Sequencing Variants To find the influence of
mutations identified in the PCDH15 on its protein structure that may
have an important role in disease susceptibility, in silico analysis was
performed. Each DNA sequence was blasted using nucleotide blast (Blastn) tools
of NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn & BLAST).
The DNA sequences were further aligned and analyzed to predict the effect of
mutation on the protein product with CLC workbench 6. The mutations identified
in PCDH15 were evaluated using online available programs to predict
whether variants are deleterious. PolyPhen classifies an amino acid substitution
as probably damaging, possibly damaging, benign, or unknown. Provean predicts
whether an amino acid substitution affects protein function. Provean prediction
tool was used, which is based on the degree of conservation of amino acid
residues in sequence alignments derived from closely related sequences,
collected through PSI-BLAST.
RESULTS
An evidence of linkage and the region of homozygosity for STR
markers at USH1F locus on chromosome 10q21.1 were observed for USH phenotype in
a consanguineous Pakistani family (Figure 1). On mutation screening of the
candidate gene, PCDH15 in this region, three sequence variants
c.1138G>A, c.1263T>C and c.1304A>C in exon 11 were identified (Table
3). However, among the three identified mutations, a c.1304A>C mutation was
found to be disease causative as it was segregating with the disease phenotype
and was also not present in 100 ethnically matched controls. This c.1304A>C
mutation predicts the substitution of an amino acid residue aspartic acid to
alanine at codon 435 (p.435D>A).
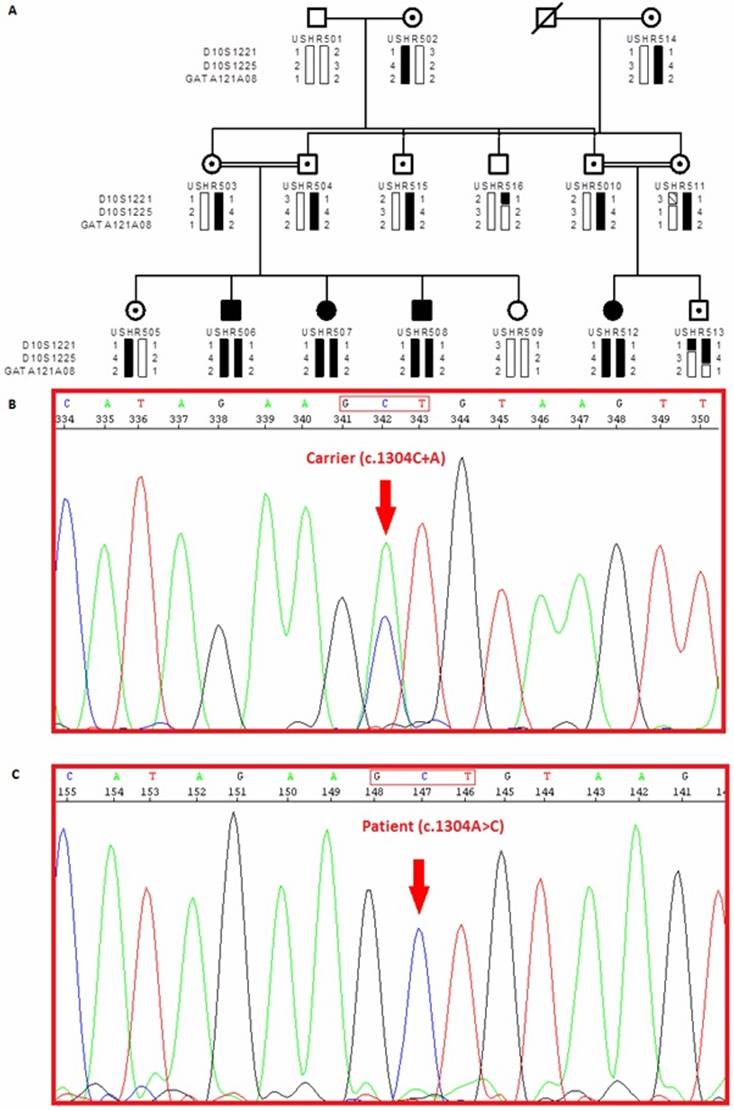
Figure 1 Pedigree of a consanguineous Pakistani family
with STR genotyping data mapped to a locus on chromosome 10q21.1 A: In pedigree individual identification
numbers are listed below the pedigree symbols. Genotyped markers from the chromosome
10q21.1 region are shown to the left, and individualsí» allele numbers for each
marker are given next to the bar. Black bars represent the haplotype
segregating with the PCDH15 gene. B: Electropherograms of PCDH15 exon 11 in a carrier individual C: Electropherograms of PCDH15 exon 11 in an affected patient; DNA sequence analysis revealed a homozygous A to C substitution at
nucleotide 1304 (from the translation start site) in the affected patient,
causing the Asp435Ala mutation.
Table 3 Mutations identified
in exon 11 of PCDH15 gene
|
Gene
name |
Accession
number |
Variant
nomenclature (coding) |
Exon
number |
Protein
change |
Classification |
|
PCDH15 |
NM_033056 |
c.1138G>A |
11 |
p.380Gly>Ser |
Polymorphism |
|
c.1263T>C |
11 |
p.421Thr>Thr |
Polymorphism |
||
|
c.1304A>C |
11 |
p.435Asp>Ala |
Pathogenic |
The multiple sequence alignment of amino acids showed that
aspartic acid at position 435 is phylogenetically conserved in different
species, and PolyPhen predicted the mutation to be possibly damaging (Figure 2). These results
suggest that aspartic acid may be functionally important and the mutation may
lead to damaging interference with conformation and function of PCDH15.
Provean prediction analysis results for p.435D>A yield a score of 3.084 and
predicted this change as deleterious. Whereas the other PCDH15
variations, p.380G>S and p.421T>T were predicted to be neutral with
Provean score of 1.860 and 0.000 respectively (Table 4).

Figure 2 In silico analysis of the PCDH15 sequencing variants A: The mutant and normal PCDH15 protein sequences were
aligned using CLC Workbench V.6 to find the difference between them. The mutant
amino acid residues, p.380Gly>Ser, p.421Thr>Thr and p.435Asp>Ala are
indicated by red arrows; B: In order to check the phylogenetic
conservation analysis of the mutated amino acid residue, amino acid sequences
of PCDH15 from human and different species were downloaded from the NCBI
and automatically aligned by Lasergene Meg Align (DNASTAR, Madison, WI,
USA).
Multiple sequence alignment indicates that asparagine (D) at position 435 (red
bar) is highly conserved; C: This diagram showing the PolyPhen
analysis results of p.435D>A mutation. The results of PolyPhen analysis
classified p.435D>A mutation as possibly damaging with a score of 0.949.
Table
4 In silico analysis of mutations identified in exon 11 of PCDH15 gene
|
Gene |
Nucleotide change |
Residual change |
Location |
PolyPhen |
SIFT |
PMut |
|||
|
Prediction |
PSIC score |
|
Score |
Prediction |
RI |
||||
|
PCDH15 |
c.1138G>A |
p.380G>S |
Exon 11 |
Benign |
1.860 |
Tolerated |
0.93 |
Neutral |
5 |
|
c.1263T>C |
p.421T>T |
Exon 11 |
Silent |
0.000 |
Tolerated |
NA |
Neutral |
NA |
|
|
c.1304A>C |
p.435D>A |
Exon 11 |
Possibly damaging |
0.949 |
Tolerated |
0.07 |
Pathogenic |
0 |
|
DISCUSSION
USH1 genetic
subtypes cannot be differentiated on the basis of clinical signs and symptoms,
only investigations of linkage analysis in linkage informative consanguineous
families[8] or mutational analysis of the genes involved,
have been considered useful. Roux et al[14]
investigated a cohort of patients in France and reported that mutations in USH1
genes cause USH in more than 90% of patients. However in some ethnic groups, a
few mutations have a significant carrier frequency. As an example, a mutation
c.216G>A in USH1C gene was reported in French Canadians of Acadian origin
that accounted for almost all USH1 cases in Acadian population[15],
but this mutation has not been found in any other population. In another
example, the mutation c.733C>T in the PCDH15 gene was identified by
Ben-Yosef et al[16], that accounted for 58% of
families of Ashkenazi with USH1. In some USH genes mutations were not found in
some sporadic and familial cases of USH from Pakistan, France and Spain,
suggested the search for additional novel USH genes[8,14,17].
The PCDH15
gene is a member of the cadherin superfamily. Family members encode integral
membrane proteins that mediate calcium-dependent cell-cell adhesion. It plays
an essential role in maintenance of normal retinal and cochlear function. The PCDH15
gene has been mapped by Alagramam et al[18]
to chromosome 10q21-q22b. Ahmed et al[12] identified
33 exons in the PCDH15 gene and spans about 1.6 Mb of human genomic DNA.
Ahmed et al[13] identified four additional exons in
the PCDH15 gene, which encode two other cytoplasmic domains. In a
Pakistani family, the first 2 exons of the PCDH15 gene were found
critical defined regions to cause USH type IF. Within the promoter region of
the PCDH15 gene instead of TATAA or CAAT sequences, a CpG island,
suppressor and enhancer elements have been identified by Alagramam et al[19].
The intron sizes in PCDH15 are variable and three additional genes have
been reported by Ahmed et al[13]. Within the PCDH15
gene, large genomic rearrangements have been found that are a significant cause
of USH1F syndrome[20].
Mutations in
the PCDH15 gene are responsible for both combined hearing and vision
impairment (USH1F) and non-syndromic deafness (DFNB23). To date, more than 30
different point mutations have been identified as well as large rearrangements,
including deletions and duplications have also been reported.
In previous
reports, the only protein truncating mutations in PCDH15 were found
associated with USH, and other substitutions were reported to be responsible
for causing DFNB23 phenotype. Ahmed et al[12] in a
consanguineous Pakistani family with USH1F, investigated the PCHD15 gene
for mutations in affected members and found a homozygous 1940C-G transversion
that resulted in a ser647-to-ter (S647X) substitution and predicted to truncate
the protein in the EC6 domain. Doucette et al[21]
reported a novel homozygous 1583T-A transversion that resulted an amino-acid
substitution of a valine with an aspartic acid at codon 528 (p.V528D) of PCDH15
in a consanguineous family from the island of Newfoundland. Ouyang et al[22]
reported the heterozygosity for a mutation of 3-bp deletion (5601-5603delAAC)
in exon 33 of PCDH15 gene as this resulted in subsequent deletion of
threonine at 1867 and this also caused a missense mutation in patient with
USH1F at PCDH15 locus. Rebibo-Sabbah et al[23]
reported nonsense mutations in patients with USH1F subsequently in translation
of a variable length protein that resulted from partial read-through of this
nonsense mutations. Zheng et al[24] also reported
patients of USH1 who carried mutations of a 1-bp deletion in the PCDH15 gene
(16delT) in compound heterozygosity with a mutation in the CDH23 gene.
The PCDH15 deletion (16delT) mutation causes a frameshift leading to an
altered amino acid sequence from codon 6, followed by a premature termination
at codon 11 in the predicted signal peptide of the protein.
In a study,
the mutation c.1304A>C (p.435D>A) identified in the PCDH15 gene by
Jaijo et al[17] in a random unrelated pool
of samples and was presumed to be non-pathogenic alteration. However the change
is clearly disease associated in our family (Figure 1). In silico analysis also
supports our findings that the change is pathogenic and conserved in different
species (Figure 2 and Table 4), which is contrary to the report published by
Jaijo et al[17].
The mutation
identified in this study occurs in a highly conserved extracellular cadherin
(EC1) domain of PCDH15 and is predicted to be more deleterious than the
previously identified missense mutations (p.R134G and p.G262D). Physical
assessment, vestibular and visual function testing in deaf adults ruled out
syndromic deafness because of USH. This study validates the DFNB23 designation
and supports the hypothesis that missense mutations in conserved motifs of PCDH15
cause nonsyndromic hearing loss. This emerging genotype-phenotype correlation
in USH1F is similar to that in several other USH1 genes and cautions against a
prognosis of a dual sensory loss in deaf children found to be homozygous for
hypomorphic mutations at the USH1F locus.
The
identification of a missense substitution mutation and establishing its
association with USH1F phenotype in a consanguineous Pakistani family is the
first example of an association of any missense mutation with USH1F. In exon 11
of PCDH15 gene, a frameshift mutation, c.1304_1305insC (p.T436YfsX12)
causing recessive USH1F was also reported by Ahmed and his colleagues[12].
It indicated that the mutations of PCDH15, affecting EC1 domain of its
protein product is responsible for causing severe phenotype, i.e. USH1F.
Recently, a novel transversion pathogenic mutation for nonsyndromic deafness in
the USH1F gene PCDH15 in a consanguineous family has been reported from
the island of Newfoundland[21]. The association of a missense
mutation and evaluation of its pathogenicity in a Pakistani consanguineous
family further support the previous studies and emphasizes the need to know the
genetic basis of recessively inherited neurological diseases in Pakistan. As a
consequence of the unique socio-cultural practices in the population of
Pakistan, approximately 60% of marriages are consanguineous, of which more than
80% are between first cousins[25]. Recessively inherited
diseases are more prevalent in population of Pakistan as cousin marriages are
common. These large consanguineous families are a powerful resource for genetic
linkage studies of recessively neurological inherited disorders. In Khyber
Pakhtunkhwa there are several factors contribute to the wide prevalence of
genetic disorders in the region including the high rate of consanguinity,
social trend to have more children until menopause, selective factors favoring
inherited disease, and the lack of public awareness towards the early
recognition and prevention of inherited disease. Different tribes of Pashtoons
living here are very intimate about their marriages of inter tribal partners
resulting in the mixing of blood and thus impurification of tribes. Many people
do not agree with medical explanations of a genetic mode of disease
inheritance, even in cases where there is an affected child. Complex
neurological disorders like USH are frequent in the population of Pakistan and
particularly in Khyber Pakhtunkhwa due to consanguinity and have a substantial
impact on health care, socio-economic level and quality of life. Finding
genetic risk factors involved in these disorders may boost knowledge about the
disorder and possible treatment therefore provide a strong background for
convincing or changing the Pakistani publicí»s view regarding cousin marriages.
In conclusion,
the evaluation of pathogenic role of mutation identified in exon 11 of PCDH15
gene and its association with USH phenotype in a consanguineous Pakistani
family will enable us to further characterize PCDH15 gene variations and
establish genotype/phenotype correlation. It will also help us to understand
genetic mechanism of disease progression. A better understanding of PCDH15
mutations and their effect on protein product and the resulting outcome that
how some mutations results into less severe phenotype (DFNB23) or more severe
phenotype (USH1). This knowledge will also be helpful in developing diagnostic
and therapeutic strategies and also help in reducing the burden of genetic
diseases by extending genetic counseling to individuals having strong history
of genetic disease/disorders.
ACKNOWLEDGEMENTS
The authors of
the article would like to thank the patients and their family members for their
help and participation in this study. We are also grateful to our other
colleagues at IBGE for their technical assistance.
Saleha S performed experimental work and paper writing, Ajmal M
participated in experimental work, Nasir M and Jamil M helped in paper writing
and formatting, Hameed A designed, analyzed data and proof read the manuscript.
All authors read and approved the final manuscript.
Foundation:
Supported
by the Kohat University of Science and Technology, Kohat, Pakistan and
Institute of Biomedical and Genetic Engineering, Islamabad, Pakistan.
Conflicts
of Interest: Saleha S, None; Ajmal M, None; Jamil M, None; Nasir M,
None; Hameed A, None.
REFERENCES [Top]
1
Bonnet C, El-Amraoui A. Usher syndrome (sensorineural deafness and retinitis
pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol 2012;25(1):42-49. [CrossRef] [PubMed]
2 Mathur P, Yang J.
Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities.
Biochim Biophys Acta 2015;1852(3):406-420. [CrossRef] [PubMed] [PMC free article]
3 Friedman TB,
Schultz JM, Ahmed ZM, Tsilou ET, Brewer CC. Usher syndrome: hearing loss with
vision loss. Adv Otorhinolaryngol
2011;70:56-65. [CrossRef]
4 Kilpinen H,
Dermitzakis ET. Genetic and epigenetic contribution to complex traits. Hum Mol Genet 2012;21(R1):R24-28. [CrossRef] [PubMed]
5 Riahi Z, Bonnet C,
Zainine R, Lahbib S, Bouyacoub Y, Bechraoui R, Marrakchi J, Hardelin JP, Louha
M, Largueche L, Ben Yahia S, Kheirallah M, Elmatri L, Besbes G, Abdelhak S,
Petit C. Whole exome sequencing identifies mutations in Usher syndrome genes in
profoundly deaf tunisian patients. PLoS
One 2015;10(3):e0120584. [CrossRef] [PubMed] [PMC free article]
6 Yan D, Liu XZ.
Genetics and pathological mechanisms of Usher syndrome. J Hum Genet 2010;55(6):327-335. [CrossRef] [PubMed] [PMC free article]
7 Ayuso C, Millan JM.
Retinitis pigmentosa and allied conditions today: a paradigm of translational
research. Genome Med 2010;2(5):34. [CrossRef] [PubMed] [PMC free article]
8 Riazuddin S,
Belyantseva IA, Giese AP, et al.
Alterations of the CIB2 calcium- and integrin-binding protein cause Usher
syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet 2012;44(11):1265-1271. [CrossRef] [PubMed] [PMC free article]
9
Millan JM, Aller E, Jaijo T, Blanco-Kelly F, Gimenez-Pardo A, Ayuso C. An
update on the genetics of usher syndrome. J
Ophthalmol 2011;2011:417217.
10 Ahmed ZM,
Riazuddin S, Khan SN, Friedman PL, Riazuddin S, Friedman TB. USH1H, a novel
locus for type I Usher syndrome, maps to chromosome 15q22-23. Clin Genet 2009;75(1):86-91. [CrossRef]
[PubMed] [PMC free article]
11
Green MR, Sambrook J. Molecular cloning.
A laboratory manual. Cold Spring Harbor Laboratory, NY: Cold Spring Harbor
Laboratory press; 2012.
12 Ahmed ZM,
Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman
TB, Riazuddin S, Wilcox ER. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet 2001;69(1):25-34. [CrossRef] [PubMed] [PMC free article]
13 Ahmed ZM,
Riazuddin S, Aye S, Ali RA, Venselaar H, Anwar S, Belyantseva PP, Qasim M,
Riazuddin S, Friedman TB. Gene structure and mutant alleles of PCDH15: nonsyndromic deafness DFNB23 and
type 1 Usher syndrome. Hum Genet
2008;124(3):215-223. [CrossRef]
[PubMed] [PMC free article]
14 Roux AF, Faugere
V, Vache C, et al. Four-year
follow-up of diagnostic service in USH1 patients. Invest Ophthalmol Vis Sci 2011;52(7): 4063-4071. [CrossRef] [PubMed]
15 Ebermann I, Lopez
I, Bitner-Glindzicz M, Brown C, Koenekoop RK, Bolz HJ. Deafblindness in French
Canadians from Quebec: a predominant founder mutation in the USH1C gene
provides the first genetic link with the Acadian population. Genome Biol 2007;8(4):R47. [CrossRef] [PubMed] [PMC free article]
16 Ben-Yosef T, Ness
SL, Madeo AC, Bar-Lev A, Wolfman JH, Ahmed ZM, Desnick RJ, Willner JP, Avraham
KB, Ostrer H, Oddoux C, Griffith AJ, Friedman TB. A mutation of PCDH15 among Ashkenazi Jews with the
type 1 Usher syndrome. N Engl J Med
2003;348(17):1664-1670. [CrossRef]
[PubMed]
17 Jaijo T, Oshima A,
Aller E, Carney C, Usami S, MillĘón JM, Kimberling WJ. Mutation screening of the
PCDH15 gene in Spanish patients with Usher syndrome type I. Mol Vis 2012;18:1719-1726. [PMC free article]
[PubMed]
18 Alagramam KN,
Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP. The mouse Ames waltzer
hearing-loss mutant is caused by mutation of PCDH15, a novel protocadherin gene. Nat Genet 2001;27(1):99-102. [CrossRef] [PubMed]
19 Alagramam KN,
Miller ND, Adappa ND, Pitts DR, Heaphy JC, Yuan H, Smith RJ. Promoter
alternative splice forms, and genomic structure of protocadherin 15. Genomics 2007;90(4):482-492. [CrossRef] [PubMed] [PMC free article]
20 Le GuĘŽdard S,
FaugĘĘre V, Malcolm S, Claustres M, Roux AF. Large genomic rearrangements within
the PCDH15 gene are a significant
cause of USH1F syndrome. Mol Vis 2007;13:102-107.
[PMC free article]
[PubMed]
21 Doucette L, Merner
ND, Cooke S, Ives E, Galutira D, Walsh V, Walsh T, MacLaren L, Cater T,
Fernandez B, Green JS, Wilcox ER, Shotland LI, Li XC, Lee M, King MC, Young TL.
Profound, prelingual nonsyndromic deafness maps to chromosome 10q21 and is
caused by a novel missense mutation in the Usher syndrome type IF gene PCDH15. Eur J Hum Genet 2009;17(5):554-564. [CrossRef] [PubMed] [PMC free article]
22 Ouyang XM, Yan D,
Du LL, Hejtmancik JF, Jacobson SG, Nance WE, Li AR, Angeli S, Kaiser M, Newton
V, Brown SD, Balkany T, Liu XZ. Characterization of Usher syndrome type I gene
mutations in an Usher syndrome patient population. Hum Genet 2005;116(4):292-299. [CrossRef] [PubMed]
23 Rebibo-Sabbah A,
Nudelman I, Ahmed ZM, Baasov T, Ben-Yosef T. In vitro and ex vivo suppression
by aminoglycosides of PCDH15 nonsense
mutations underlying type 1 Usher syndrome. Hum
Genet 2007;122(3-4):373-381. [CrossRef] [PubMed]
24 Zheng QY, Yan D,
Ouyang XM, Du LL, Yu H, Chang B, Johnson KR, Liu XZ. Digenic inheritance of
deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15
in mice and humans. Hum Mol Genet
2005;14(1):103-111. [CrossRef]
[PubMed] [PMC free article]
25
Perveen F, Rehman S. Consanguineous marriages and their malformation in F1
Generation. Asia J Pharma Health Sci
2012;2(3):406-411.
[Top]