·Review··Current Issue· ·Achieve· ·Search Articles· ·Online Submission· ·About IJO·
Characteristics of corneal dystrophies:
a review from clinical, histological and genetic perspectives
Ze-Nan Lin, Jie Chen, Hong-Ping Cui
Department
of Ophthalmology, Shanghai East Hospital, Tongji University School of Medicine,
Shanghai 200120, China
Correspondence to:
Hong-Ping Cui. Department of Ophthalmology, Shanghai East Hospital, Tongji
University School of Medicine, Shanghai 200120, China. hpcui@vip.163.com
Received: 2015-07-19
Accepted: 2015-08-16
Abstract
Corneal
dystrophy is a common type of hereditary corneal diseases. It includes many
types, which have varied pathology, histology and clinical manifestations.
Recently, the examination techniques of ophthalmology and gene sequencing
advance greatly, which do benefit to our understanding of these diseases.
However, many aspects remain still unknown. And due to the poor knowledge of
these diseases, the results of the treatments are not satisfoctory. The purpose
of this review was to summarize the clinical, histological and genetic
characteristics of different types of corneal dystrophies.
KEYWORDS: corneal
dystrophy; clinic; histology; gene mutation
DOI:10.18240/ijo.2016.06.20
Citation: Lin ZN, Chen J, Cui HP. Characteristics of corneal
dystrophies: a review from clinical, histological and genetic perspectives. Int J Ophthalmol 2016;9(6):904-913
INTRODUCTION
Corneal dystrophies (CDs) are a group of commonly-occurring primary,
progressive corneal diseases. Depending on the anatomical sites, CDs can be
classified into 3 subtypes: 1) anterior CDs include anterior basement membrane
dystrophy (ABMD) and MeesmanˇŻs epithelial dystrophy; 2) stromal CDs include
Reis-BuecklerˇŻs dystrophy, honeycomb dystrophy, lattice dystrophy, granular
dystrophy, Avellino dystrophy, macular dystrophy, Schnyder crystalline
dystrophy, Fleck dystrophy, and congenital hereditary stromal dystrophy; 3)
endothelial CDs include FuchˇŻs dystrophy, congenital hereditary endothelial
dystrophy, and posterior polymorphous dystrophy. Most CDs are characterized
with varied shapes of corneal opacities. CDs have been investigated by many
ophthalmologists worldwide, but their mechanisms remain unclear in many cases.
Although CDs are still enigmatic, our knowledge about them has been expanded
greatly in recent years due to the development of gene sequencing techniques
and ophthalmological examination advances [e.g.
high-definition optical coherence tomography (OCT), confocal microscopy]. This
review highlights the advances in our understanding of CDs based on researches
in recent years.
MATERIALS
AND METHODS
Search Strategy To identify the relevant studies, we
searched PubMed for papers investigating the clinical manifestations, histology
or genetics of CDs. The search was through May 2015 with no language
restrictions. Each subtype of CDs was searched separately. For example, the
searching key items of ABMD include: ABMD, epithelial basement membrane dystrophy (EBMD), Cogan microcystic dystrophy, map-dot-fingerprint (MDF) dystrophy,
clinic, histology, genetics, gene mutation. The other CDs were searched with
the same means. In addition, we manually reviewed the reference lists from the
relevant articles.
Study Selection We aimed to identify all the relevant
studies that investigate the clinical, histological or genetic aspects of each
types of CDs. We applied the following exclusion criteria: 1) editorials or
letters; 2) case series or case reports; 3) studies not investigating the
clinic, histology or genetics of CDs; 4) studies not conducted in humans or
mice. The first two authors independently reviewed all searched results to get
the eligible articles. Discrepancies between the two authors were resolved by
the consensus of the third author of this review. In the end, we identified 99
relevant articles, which were used as reference articles in our review.
Anterior (Epithelial and BowmanˇŻs
Membrane)
Anterior basement membrane
dystrophy ABMD
[online mendelian inheritance in man (OMIM) 121820] is also known as EBMD, Cogan microcystic dystrophy or
MDF dystrophy. ABMD is characterized by
subepithelial bleb-like microcysts, fingerprint lines, geographic map-like
lines, and epithelial microcysts or dots, which are all bilateral and
frequently asymmetric, revealed by slit-lamp examination. About 10% of ABMD patients develop painful recurrent epithelial erosions[1]. The cause of ABMD remains controversial. Though it is more likely to be
age-related, the hereditary pathways in some cases are seemingly autosomal
dominant or X-chromosome-related[2-3].
ABMD is
histologically characterized by the thickened
epithelial basement membrane (EBM) which duplicates and/or insinuates into the
corneal epithelium, and the presence of hyperreflective dots, which result in
the classical manifestation of MDF opacities in the cornea on slit-lamp
examination. More recently, a more finest ultrastructure of ABMD in some cases was studied with confocal microscopy[4] and
standard-definition (SD)-OCT[2].
The ABMD lesions have variable shapes (e.g. map-, dot-, fingerprint- or bleb-like). In the
superficial/basal epithelium and BowmanˇŻs membrane under microscopy, the
map-like lesion of the cornea presents a different shape of high-reflective
extracellular deposits, while the fingerprint-like lesion presents multiple
dark striae[4]. Both
lesions show a thickened EBM, which invaginates into the epithelium in the form
of multi-sheet fibrogranular material[5-8].
The dot-like lesion has 2 subtypes: Cogan cysts and the cysts reported by Bron
and Brown[7]. Cogan cysts are the cell
degeneration products that aggregate in the form of cyst underneath an
intraepithelial sheet. The second subtype is a sheet of fibrogranular material
in the EBM and BowmanˇŻs membrane. Besides the classical MDF type, there are
also some other subtypes, such as Band-shaped
and whorled microcystic dystrophy. Under light microscopy, the scraped
epithelium shows a transition of normal corneal epithelium into the zone where
the cytoplasm is distended with abundant fine vacuoles. Swollen cells are
present at all levels of epithelium, and neither periodic acid-Schiff (PAS) nor
Alcian blue acid mucopolysaccharide stain shows cytoplasmic positivity[3].
Gelatinous
drop-like dystrophy (GDLD) (OMIM 204870), or
familial subepithelial corneal amyloidosis, which has an autosomal recessive
hereditary pattern, was first reported in 1914[9]. Though the incidence rate was about 1 in 30 000 in
Japan, it is very rare in other countries[10].
GDLD is characterized by an accumulation of amyloid substances in the
subepithelial region of the cornea, which have several shapes (yellowish-white,
mulberry-like, gelatinous) (Figure 1). In the first decade of life, the
accumulation of these substances leads to vision disturbance, foreign-body
sensation, photophobia, and lacrimation. In the later stages, neovasculation
may occur in the subepithelium and superficial stroma[11]. Surgical intervention is temporally effective, but
recurrence within a few years has been reported[12-13].
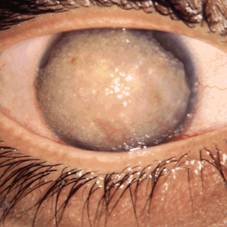
Figure 1 Gelatinous drop-like dystrophy, courtesy of Dr.
GK Klintworth.
GDLD is correlated with the gene mutations on the tumor-associated calcium
signal transducer 2 (TACSTD2)[14].
The TACSTD2-encoded protein is a monomeric cell surface glycoprotein expressed
in the cornea, trophoblasts, and most carcinomas[15-16]. To date, more than 20 mutations in TACSTD2 have
been identified[11].
In Japan, the major mutation identified in GDLD cases is Q118X in TACSTD2[17].
Histological biopsy reveals a thinned corneal epithelium with an
incompletely destroyed BowmanˇŻs membrane and subepithelial and stromal amyloid
deposits, partially arranged in a band-shape[12]. Immunohistochemical and proteomic analyses reveal
that amyloid fibril formation may be attributed to abnormal accumulation of
lactoferrin and transforming growth factor beta-induced protein (TGFBIp) [18-19].
Amyloid nodules in the subepithelial layer and the anterior corneal stroma are
stained with Congo red to form apple-green birefringence when combined with
polarized light[20].
Despite the discovery of many gene mutations, the mechanism of amyloid
formation remains unclear[11].
As reported, the abnormal proteins found in the amyloid lesions of GDLD are
rich in advanced glycation end (AGE) products and D-b-aspartic acid. It is
proposed that the amyloid fibril formations in GDLD may be caused by the
non-enzymatic post-translational modifications of proteins, including AGE
formation and isomerisation of aspartyl residues[21].
MeesmannˇŻs
epithelial corneal
dystrophy MeesmannˇŻs
epithelial corneal
dystrophy (MECD) (OMIM 122100) is a rare bilateral disorder
confined to the corneal epithelium. Its symptomatic intraepithelial microcysts
appear in the first few years of life and can be seen under a slit lamp[22]. Under slit-lamp
biomicroscopy, the lesions appear as punctate, bubble-like, round or oval
opacities in the corneal epithelium[23].
Nevertheless, vision is usually not affected[24]. MECD is mostly considered as an autosomal dominant
inherent disease, but an autosomal recessive form is also reported[23]. MECD has been
linked to gene mutation in K3 and K12, which are expressed in the corneal
epithelium[25-26].
The dystrophic epithelium is histologically characterized by cellular
swelling, cyst-like inclusions, and cytoplasmic vacuoles. The cysts contain
PAS-positive degenerated cell debris[27]
and are a dense intracellular substance of unknown composition[28]. Electron
microscopy has revealed an electron-dense and amorphous ˇ°peculiar substanceˇ± in
the cytoplasm of epithelial cells. Deposition of the peculiar substance in the
epithelium leads to cyst formation and cell death, followed by rapid epithelial
regrowth[27].
Stromal Corneal Dystrophies
Reis-B¨ąckler corneal dystrophy Reis-B¨ąckler
corneal dystrophy (RBCD) (Corneal Dystrophy of BowmanˇŻs I, CDB1,
OMIM 608470) was first reported in 1917 by Reis
and elaborated in 1949 by B¨ącklers[29-30].
The
affected patients experience recurrent painful
erosions of corneal epithelium within the first few years and moderate
impairment of visual loss. With aging, however, map-like and ring-like
opacities appear in BowmanˇŻs membrane, and these lesions become denser and
irregular. After the second decade, patients may feel less pain due to the
decrease of corneal sensitivity[31].
RBCD is associated with the R124L mutation in transforming growth factor,
beta-induced (TGFBI) gene or with atypical cases of F540, H626R, G623D or R124C
mutations[32]. However,
there are rare reports on RBCD in Chinese patients.
The materials in the opacities are eosinophilic, congophilic and are not stained with
PAS on histopathological examination[30].
Light microscopy reveals rod-shaped and trapezoidal deposits in the BowmanˇŻs
layer and between epithelial cells[32].
This pathological finding is consistent with the superficial granular dystrophy[31].
Avellino Dystrophy Avellino
dystrophy, also known as Granular corneal dystrophy type II (GCD2, OMIM
607541), was first reported in patients from Avellino, Italy[33]. GCD2 belongs to
the stromal CDs, which also include GCD and lattice corneal dystrophy (LCD).
The classical manifestations of GCD2 combine the characteristics of GCD and LCD
with discrete granular and lattice opacities. The granular opacities appear
earlier and more commonly than the lattice opacities[34]. The opacities could lead to the disturbance of
visual acuity, but their location and severity decide the final outcomes[34]. The onset seems
to be earlier in homozygote than in heterozygote patients[35]. GCD2 is associated with Arg124His mutation in
TGFBI, mapped to chromosome 5q, and has
an autosomal dominant pattern[34]. It is proposed that
the pathogenesis of GCD2 may be critically related to defective autophagy[36].
However, its mechanism is still poorly understood.
Histologically,
GCD2 patients have both hyaline granular deposits, which are located
superficially, and amyloid lattice deposits, which appear at deeper sites[35]. The hyaline
granules and amyloid lattice lines are stained with Congo red and MassonˇŻs
trichrome, respectively. Depending on the shapes, GCD2 lesions can be divided
into 3 types: 1) type 1, diffuse hazy deposits are superficially
located in an irregular soft pattern; 2) type 2, granular deposits are
subdivided into superficial round granular deposits (type 2a) and superficial
round spiculated ones (type 2b). In GCD2 linear (lattice) deposits, the
branches radiated out from the main deposit or trunks are well below the Bowman
layer and appear dense and white; 3) some deposits have short side branches
(type 3a, <trunk width), while others have long side branches (type 3b, >
trunk width) [34].
Central
cloudy dystrophy of Francois Central
cloudy dystrophy of Francois (CCDF), first
reported in 1955[37],
is characterized by polygonal cloudy gray stromal opacities separated by
relatively clear lines, which creates a leather-like crocodile appearance in
the central cornea. Under the slit lamp, CCDF is larger and more numerous in
the posterior part of the stroma and becomes smaller and less frequent in the
anterior part. The anterior layers are unaffected in some cases, but the grey
patches reach the Bowman's membrane in other cases. The corneal endothelium and
epithelium are unaffected[38].
This condition is presumably an autosomal dominance, but its detailed mechanism
is unknown[39]. In
contrast, similar corneal opacities located at either the central or peripheral
cornea in the deep stromal layer are known as ˇ°posterior crocodile shagreenˇ±
and are usually considered as age-related corneal degenerations. The
distinction between two entities is an inheritant pattern[40].
Histologically,
light microscopy reveals stromal staining for acid mucopolysaccharide [39]. Transmission
electron microscopy (TEM) identifies extracellular vacuoles, some of which have
fibrillogranular substances and electron-dense deposits. The opacities result
from the extracellular accumulation of mucopolysaccharide and lipid-like
material[39].
SchnyderˇŻs central crystalline
dystrophy SchnyderˇŻs
central crystalline dystrophy (SCCD) (OMIM
121800), first described by Schnyder and van Went, has an autosomal dominant
inherited pattern. SCCD is characterized
by a bilateral clouding of the central cornea, arcus lipoides and/or visible
crystalline deposits of cholesterol in the stroma. There is accumulation of
phospholipid, unesterified cholesterol and cholesterol ester in the corneal
stroma[41]. The precise mechanism remains unclear. Gene
mutations are considered to be localized at 1p34.1-p36 interval and in some
candidate genes: FABP3, CTPS, SCP2, COL8A2, GALE
and MTHFR [41].
To date, mutations of UBIAD1 have been identified in 28 unrelated
families with SCCD[42-45].
Common systemic findings associated
with SCCD include hypercholesterolemia and hyperlipidemia, but their presence
is not mandatory for the pathogenesis of SCCD.
Histologically, diagnosis of SCCD is confirmed by the lipid and
cholesterol deposits in the corneal stroma on oil red O staining under corneal
biopsy. Under in-vivo confocal
microscopy, superficial epithelial cells appear in normal limits, while the
basal cell layer is poorly visualized and presents crystalline deposits
extending from the anterior stroma. Moreover, large or multiple deposits of
brightly reflective crystalline material extend from the anterior stroma to the
middle part, while the regularity and density of keratocytes are remarkably
decreased. Although poorly visualized because of the increased reflectivity of
the anterior cornea, the posterior stroma shows fine needle-shaped deposits in
the posterior stromal matrix, but number decreases with depth and the
brightness is reduced compared with the deposits in the anterior stroma[46].
Congenital
stromal corneal dystrophy Congenital
stromal corneal dystrophy (CSCD) (OMIM
610048) is very rare. Its clinical manifestations include the diffused,
bilateral and corneal clouding of flake-like whitish opacities throughout the
stroma. The lesions appear shortly after birth and progress with age. Some
affected patients also suffer from strabismus or nystagmus. Most patients
undergo a penetrating keratoplasty in early adulthood with good outcomes[47]. CSCD is the only
known human disease associated with the mutated gene of decorin, a small
leucine-rich proteoglycan (SLRP)[48].
Decorin is involved in the control of fibrillogenesis and fibril organization,
which contribute to corneal transparency and refractive stability[49]. It is proposed that a truncated SLRP
protein core is retained and accumulates intracellularly. This process triggers
endoplasmic reticulum stress, which leads to abnormal synthesis and secretion
of SLRP and ultimately to impairment of stromal structure and corneal
transparency [48].
Histologically, epithelial cells are normal under confocal microscopy, but
the reflectivity is increased throughout the stromal layer. In CSCD patients,
the lamellar stromal structure is disrupted, which is more severe in the
anterior and posterior central stroma. The Fourier-domain OCT images also show
higher diffuse reflectivity in the stroma than in the normal cornea. Under
electron microscopy, the electron-lucent zones in the corneal stroma are
located between the normal lamellae of collagen fibrils with thinned filaments
in haphazard arrangement[47].
Francois-Neetans Fleck corneal dystrophy Fleck corneal dystrophy (FCD), also called Francois-Neetens FCD (OMIM 121850), is very rare and
first described in 1956[50].
Slit-lamp examination reveals bilateral, flat, gray-white, oval or round
discrete opacities throughout the corneal stroma. No systemic
abnormality has been reported[51].
FCD occurs early in life but then progresses slowly, and visual acuity is not
greatly disturbed. Thus, treatment is not necessary in most cases. Recurrence
was not reported in a 10-year follow-up after penetrating keratoplasty[52]. FCD is caused by
mutations in PIP5K3 and has an autosomal dominant pattern[53-54]. PIP5K3 gene is responsible for intracellular
accumulation and engorgement and the reported mutations result in truncation of
PIP5K3 protein before its structure is formed, leading to the abnormal activity
of PIP5K3 protein. Further studies are needed to elucidate the function of
PIP5K3 protein in FCD patients and normal persons[55].
Histopathologically, some keratocytes contain fibrillogranular material in
relatively large intracytoplasmic vacuoles, while some keratocytes contain
pleomorphic electron-dense and membranous intracytoplasmic inclusions[56]. The materials
were predicted to contain lipids and acid mucopolysaccharides[54].
GCD I (Groenouw type I) belongs to the TGFBI-associated
CDs, which also include RBCD, Thiel-Behnke corneal dystrophy (TBCD), LCD, and
GCD II. GCD I is characterized by the discrete opacities in the corneal stroma,
which are irregularly crum- or flake-like and appear slightly whitish or glassy
(Figure 2). Though most patients are asymptomatic, some patients develop
recurrent erosions. The lesions become more numerous and severe with time,
leading to visual acuity impairment. Some patients may require keratoplasty in
the fifth decade or later[31]. GCDI is
autosomal dominant and associated with the mutations on TGFBI. The predominant
one of the varied mutations is Arg555Trp[57]. The
Arg555Trp mutation will lead to the abnormal degradation/turnover of corneal
TGFBIp, and finally to the accumulation and increased propensity to aggregate
through electrostatic interactions[58].
Histologic findings with Masson Trichrome red staining and without Congo
red staining were tested (Figure 3). Electron microscopy shows eletron-dense
rod-like deposits and microfibrils in keratocytes and epithelial cells. The
materials are ascribed to phospholipid[31]. Though
the lesions affect mostly the stroma, they do occur within the whole depth of
the cornea in some cases.
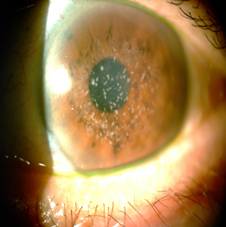
Figure 2 Granular corneal dystrophy (Groenouw
type I).

Figure 3 Light
microscopy of GCD I. Masson Trichrome stain, courtesy of Dr. GK Klintworth.
Lattice corneal dystrophy LCDs are a
subgroup of stromal CDs. All LCDs have amyloid accumulation in the stroma and
are often arranged in a branching pattern[31].
LCDs have an autosomal dominant pattern and is related to the mutations on the
TGFI gene, which encodes keratoepithelin, an extracellular matrix that mediates
cell adhesion[59]. LCDs are
classified into 3 subtypes: LCD I (OMIM 122200), LCD ˘ň (OMIM 105120), and LCD ˘ó and LCD ˘óA
(OMIMs 204870 and 608471). LCD I is most commonly-seen. The abnormalities of
LCD I occur in the first or second decade of life and progress over time. Its
anterior stroma has rod-like or linear opacities. Recurrent erosions are common
and central anterior stromal haze may develop with age. The lesions usually
affect the anterior and central corneas, leaving a relatively normal periphery
cornea. The mutation at codon 124 of TGFBI, where the amino acid arginine is
replaced by cysteine, is previously considered as the most frequent defect of
LCD I. LCD ˘ň is
associated with systemic amyloidosis type V (Meretoja syndrome/Finnish type),
which is an autosomal dominant systemic disease. LCD ˘ň occurs in the early adulthood and
affects cornea, skin, and cranial nerves[60]. LCD ˘ó is
clinically manifested as the presence of thick ropy lattice lines in the cornea
compared with other subtypes. LCD ˘ó has
an autosomal recessive inheredance pattern and rare corneal erosions. Its
lesions appear usually in the fourth decade of life. LCD ˘ó A
has almost the same changes, except that it has recurrent erosions and a
dominant inheritance pattern[31].
Histologically, LCD I is considered to affect both the stroma and the
epithelium[61]. The stroma
show dense deposits, which can be stained with Congo red, PAS, and MassonˇŻs
trichrome. Dichorism and birefringence appear under polarized light, and
fluorescence occurs with thioflavin-T[31]. In LCD ˘ň, the amyloid
marker, Congo red, will show material deposition under BowmanˇŻs layer and
sometimes at the EBM level[60].
Histopathologically, amyloid deposits of LCD ˘ó are located in the
middle and superficial stromata beneath the BowmanˇŻs membrane[31]. In LCD ˘ó A, the deposits
can be stained by Congo red and display red-apple green birefringence under
polarized light, thus showing an amyloid component. They are also stained
partially positive with Masson trichrome, suggesting the presence of hyaline
components in the deposits. The lattice deposits are immunoreactive with the
anti-TGFBIp antibody. The epithelium shows dehiscence from the Bowman layer. No
abnormality is found in DescemetˇŻs membrane (DM) or the endothelium[62].
Macular corneal
dystrophy Macular
corneal dystrophy (MCD) (OMIM 217800) is very rare and has an autosomal recessive
inheritance pattern. Its prevalence rate varies among different countries[63]. MCD begins in
early years of life with superficial gray-white opacities concentrated in the
middle cornea. With aging, the lesions spread to the periphery and involve the
entire corneal stroma. Another characteristic manifestation is corneal thinning[64]. The opacities and
abnormal structure of the cornea can lead to severe visual impairment[65]. MCD is
associated with the mutations on the CHST6 gene, which encodes corneal
N-acetylglucosamine 6-O-sulfotransferase (C-GlcNAc6ST), an enzyme that
transfers sulfate to the unsulfated keratan chains on lumican. Lumican helps to
maintain the crucial size and ordered structure as well as corneal
transparency. It also influences corneal hydration and therefore corneal
transparency[66]. MCD can be
classified into subtypes I and II, defined by the absence or presence of
sulfated keratan sulfate (KS) in the serum. A third subtype, type IA, with KS
present in the keratocytes but absent in the cornea and the serum, has been
described in MCD patients from Saudi Arabia[65].
Histologically, the cornea in MCD is characterized by the accumulation of
extracellular deposits in the stroma and DM as well as by intracellular storage
of similar material in the keratocytes and corneal endothelium. The deposits
stain with Alcian blue and other histochemical methods for glycosaminoglycans
(GAGs). Biochemical studies based on organ cultures of corneas as well as serum
analyses of MCD patients suggest that the basic defect in MCD lies in a
sulfotransferase, which is specific for sulfation of KS proteoglycan. Molecular
genetic studies on MCD contribute to the mapping of the MCD gene and then to
the identification of the carbohydrate 6-sulfotransferase (CHST6) gene,
which codes corneal N-acetyl glucosamine 6-sulfotransferase, as the cause for
MCD[67].
Pre-DescemetˇŻs corneal dystrophy Pre-DescemetˇŻs
corneal dystrophy (PDCD), or deep filiform dystrophy, is very rare. PDCD was
first described and called as cornea farinata in 1923[68]. To date, little research is done in PDCD. It is
characterized with fine morphological opacities in the posterior stroma. The
lesions are composed of lipids[69].
PDCD is age-related, but the pathology remains unclear[70]. The affected patients are usually asymptomatic, and
their visual acuity is very rarely affected[71].
The onset time is usually the fourth to seventh decade. PDCD can be subdivided
into deep filiform dystrophy, deep punctiform dystrophy, polychromatic
dystrophy, and corneal farinata. These dystrophies have similar essential
characteristics, but they differ in color under direct and indirect slit-lamp
illumination. Since the deposits are uniform throughout the cornea, they
present a variety of colors that are constant[68].
Histopathologic examination of one PDCD patient demonstrates that the pathologic findings are
limited to the keratocytes of the posterior stroma[72]. The keratocytes are cytoplasmic vacuoles containing
lipid-like materials, which on electron microscopy consist of fibrillogranular
and electron-dense lamellar inclusions. No extracellular deposition of a
similar material was noted. These findings suggest that the accumulated materials
are most likely lipofuscin, a degenerative pigment that accumulates in aged
cells[70].
Posterior Corneal Dystrophies
Congenital hereditary endothelial
dystrophy Congenital
hereditary endothelial dystrophy (CHED) is a rare
inherited disorder of the corneal endothelium and characterized by corneal
opacification and nystagmus. The onset time of CHED is usually at birth and
shortly thereafter. The malfunction and degeneration of the corneal endothelium
lead to corneal edema, especially the stroma, and make the cornea appear as
ground glass. The condition is known to occur in two genetic forms: autosomal
dominant (CHED1, OMIM 121700) and autosomal recessive (CHED2, OMIM 217700).
CHED1 is more rare and has some clinical similarities with the posterior polymorphous
dystrophy (PPMD)[73],
while CHED2 is more severe and usually more common. CHED1 and CHED2 have been
mapped to chromosome 20 at two distinct loci: 20p11.2-q11.2 for CHED1[74] and 20p13 for
CHED2[75].
Histopathological analysis identified a markedly thickened DM and an
atrophied endothelium in CHED2 patients. Additionally, the patientˇŻs cornea
also had amyloid deposition and spheroidal degeneration. The presence of
amyloid was confirmed based on the presence of apple green birefringence viewed
under a polarizing filter[76].
Fuchs endothelial corneal dystrophy (FECD) (OMIM 136800)
is the most common type of endothelial CD. Bilateral, non-inflammatory and
progressive loss of endothelium results in visual loss. FECD is characterized
by guttata within cornea, stromal edema, and microcystic epithelial edema
(Figure 4). The primary defect is corneal endothelial degeneration, and the
secondary defect is corneal edema. Associated manifestations include prominent
corneal nerves, stromal opacification, recurrent corneal erosions, open angle
glaucoma, female gender, and familial predisposition[77].
Most cases are sporadic, and autosomal dominant inheritance has been recognized
in familial cases[78-79]. In
summary, mutations in FECD have been found in two transcription factors
(TCF4/E2-2 and TCF8/ZEB-1), one collagen subunit (COL8A2), and two membrane
proteins (LOXHD1 and SLC4A11/NaBC1). Except LOXHD1, these mutations appear to
converge on the collagen secretion and water pump functions of corneal
endothelium[80].
Histologically, some endothelial cells (ECs) are assumed to be
fibroblast-like, including swollen mitochondria, dilated endoplasmic reticulum
with granular material, increased number of cytoplasmic filaments, and
phagocytosed pigment granules[81-83], especially
when the posterior fibrillar layer (layer 4) of DM coveres the guttae in the
posterior banded layer (layer 3)[83]. TEM and
SEM present microvilli, increased number of hemi-desmosomes, and the positive
immune-labelling of pancytokeratin and cytokeratin-7, which are markers usually
present in almost all cells of epithelial origin[82].
Some FECD specimens had ECs with extremely long filopodia up to 100 mm long
that were immuno-positive for KS and orientated in the same plane, giving the
impression of mass cell migration in one direction[84] (Figure 5).
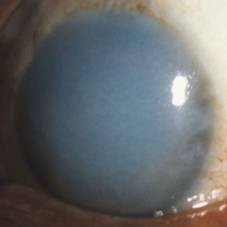
Figure 4 Fuchs
endothelial corneal dystrophy, courtesy of Dr. GK Klintworth.

Figure 5 Light
microscopy of FECD, PAS stain. Courtesy of Dr. GK Klintworth.
Posterior polymorphous corneal dystrophy Posterior polymorphous corneal dystrophy (PPCD) is a rare corneal disease
and mainly affects the DM and the corneal endothelium. PPCD is characterized by
the asymmetric
patches of grouped vesicles, scalloped bands, geographic gray hazy areas, and
epithelial-like endothelium (loss of contact inhibition with proliferation and
growth over angle and iris). Some patients may develop stromal edema. Moreover,
iris and pupil may change similarly to those in iridocorneal endothelial
syndrome. Broad peripheral anterior synechiae and glaucoma are also common.
Patients may have symptoms of pains, foreign body sensation, tearing,
photophobia, and decreased vision. PPCD is
classified into 3 subtypes: PPCD1 (OMIM 122000), PPCD2 (OMIM 609140), and PPCD3
(OMIM 609141), which are associated with the gene mutations on VSX1, COL8A2 and
ZEB1, respectively[85-89].
Histologically, the lesions of PPCD are located at the level of the
endothelium and DM. The lesions have 3 types: vesicle-type lesions, band
lesions and diffuse opacities. The former 2 types are more common than the last
one[90-95]. Under
slit-lamp, vesicles appear as blister or bleb-like, with an optically clear
centre and a small halo of grey-white haze[93].
Previous studies show ˇ°epithelium-likeˇ± multilayered cells scattered in areas
of normal endothelium and deposition of abnormal collagen material on DM,
forming an abnormal posterior collagenous layer[90,94,96-97]. The 4 types of cells shown on
the posterior corneal surface in PPCD are normal ECs, attenuated or degenerating
ECs, fibroblast-like cells, and epithelial-like cells[95].
DISCUSSION
CDs include many subtypes. While some are age-related corneal diseases,
most of them are associated with the gene mutations. They are different in
causes, clinical manifestations, development, treatments and diagnosis.
The most common symptom may be visual loss. It appears in many types of
CDs, such as ABMD, GDLD, RBCD, Avellino CD, GCD, MCD. Nevertheless, the
severity of each type may be different, for example, MCD patients may suffer
blindness with aging, which requires keratoplasty, the other patientsˇŻ (PPCD,
FCD etc.) visual acuity remains quite
stable in the most part of their lifes. Besides visual loss, foreign body
sensation, recurrent erosions, lacrimation and photophobia are also commonly
seen in patients. Some other symptoms like strabismus, nystagmus, glaucoma and
synechiae are rare, but they may appear in PPCD or FECD.
Histological experiments done on the CDs also present varied findings. In
most CDs, the lesions would lead to an abnormal corneal structure, especially
in the lesions-concentrated field. However, the corneal structure shows no
obvious changes in some types of CDs like CSCD. The lesions may locate in the
cell or extracellular space. The substances of the lesions of different types
of CDs may be lipid-like materials, acid mucosaccharide or abnormal proteins,
which have distinct results after staining with PAS, Masson Trichrome, Congo
red etc.
Thanks to the advances of gene sequencing techniques in recent years, more
and more gene mutations associated with the CDs are identified. The mutated
genes of CDs include: VSX1, COL8A2, ZEB1, TCF4/E2-2, TCF8/ZEB-1, COL8A2,
LOXHD1, SLC4A11/ NaBC1, CHST6, TGFBI, PIP5K3, UBIAD1, K3, K1, TACSTD2 etc.
According to the findings of histology and gene sequencing, the hypothesis of
some special CDs were proposed by ophthalmologists. For example, Underhaug et al[58]
thought that the gene mutations of the TGFBI may result in reduction of the
proteolytic susceptibility of the mutated TGFBIp leading to the abnormal
depositions of the TGFBIp in GCD. Morever, Choi et al[36] considered
that autophagy may play an important role in the accumulation of the TGFBIp in
GCD. However, the exact detailed mechanisms of most CDs remain unclear.
Because of the poor understanding of CDs, there are no efficient treatment
methods. Among the treatments, keratoplasty is the ultimate chance to improve
the visual acuity of severe patients. However, many patients undergoing
keratoplasty may suffer from recurrence within a few years. Because the gene
mutations play an important role in most types of CDs, gene research would
greatly contribute to the understanding of CDs, leading to a new evolutionary
treatment method. In recent years, in-vitro
gene therapy experiments have already been done [98]
and the results give us a perspective vision to cure CDs.
ACKNOWLEDGEMENTS
Conflicts of
Interest:
Lin ZN, None; Chen J, None; Cui HP,
None.
REFERENCES
1 Laibson PR, Krachmer JH.
Familial occurrence of dot (microcystic), map, fingerprint dystrophy of the
cornea. Invest Ophthalmol Vis Sci 1975;14(5):397-399.
2 El Sanharawi, Sandali O, Basli E, Bouheraoua N,
Ameline B, Goemaere I, Georgeon C, Hamiche T, Borderie V, Laroche L.
Fourier-domain optical coherence tomography imaging in corneal epithelial
basement membrane dystrophy: a structural analysis. Am J Ophthalmol 2015;159(4):755-763. [CrossRef] [PubMed]
3 Charles NC, Young JA, Kumar A, Grossniklaus HE,
Palay DA, Bowers J, Green WR. Band-shaped and whorled microcystic dystrophy of
the corneal epithelium. Ophthalmology 2000;107(9):1761-1764. [CrossRef]
4 Kobayashi A, Yokogawa H, Sugiyama K. In vivo
laser confocal microscopy findings in patients with map-dot-fingerprint
(epithelial basement membrane) dystrophy. Clin
Ophthalmol 2012;6:1187-1190. [CrossRef]
[PubMed] [PMC free article]
5 Cogan DR, Kuwabara T, Donaldson DD, Collins E.
Microcysticdystrophy of the cornea: a partial explanation for its pathogenesis.
Arch Ophthalmol 1974;92(6):470-474. [CrossRef]
6 Laibson PR. Microcysticcornealdystrophy. Trans Am Ophthalmol Soc 1976;74:488-531.
[PMC free article] [PubMed]
7 Bron AJ, Brown NA. Some superficial corneal
disorders. Trans Ophthalmol Soc U K 1971;91:XII+. [PubMed]
8 Rodrigues MM, Fine BS, Laibson PR, Zimmerman
LE. Disorders of the corneal epithelium: a clinicopathologic study of dot,
geographic, and fingerprint patterns. Arch
Ophthalmol 1974;92(6):475-482.
[CrossRef]
9 Nakaizumi G. A rare case
of corneal dystrophy. Acta Soc Ophthal
Jpn 1914;18:949-959.
10 Weber FL, Babel J. Gelatinous drop-like
dystrophy. A form of primary corneal amyloidosis. Arch Ophthalmol 1980;98(1):144-148. [CrossRef]
11 Paliwal P, Gupta J, Tandon R, Sharma N,
Titiyal JS, Kashyap S, Sen S, Kaur P, Dube D, Sharma A, Vajpayee RB.
Identification and characterization of a novel TACSTD2 mutation in gelatinous
drop-like corneal dystrophy. Mol Vis 2010;16:729-739. [PMC free article] [PubMed]
12 Uhlig CE, Groppe M, Busse H, Saeger W.
Morphological and histopathological changes in gelatinous drop-like corneal
dystrophy during a 15-year follow-up. Acta
Ophthalmol 2010;88(7):e273-374.
[CrossRef] [PubMed]
13 Quantock AJ, Nishida K, Kinoshita S.
Histopathology of recurrent gelatinous drop-like corneal dystrophy. Cornea 1998;17(2):215-221. [CrossRef]
14 Tsujikawa M, Kurahashi H, Tanaka T, Nishida K,
Shimomura Y, Tano Y, Nakamura Y. Identification of the gene responsible for
gelatinous drop-like corneal dystrophy. Nature
genetics 1999;21(4):420-423. [CrossRef]
[PubMed]
15 Ripani E, Sacchetti A, Corda D, Alberti S.
Human Trop-2 is a tumor associated calcium signal transducer. Int J Cancer 1998;76(5):671-676. [CrossRef]
16 Stein R, Basu A, Chen S, Shih LB, Goldenberg
DM. Specificity and properties of Mab RS7-3G11 and the antigen defined by this
pancarcinoma monoclonal antibody. Int J
Cancer 1993;55(6):938-946. [CrossRef]
17 Tsujikawa M. Gelatinous drop-like corneal
dystrophy. Cornea
2012;31(Suppl.1):S37-S40. [CrossRef] [PubMed]
18 Akhtar S, Bron AJ, Qin X, Creer RC, Guggenheim
JA, Meek KM. Gelatinous drop-like corneal dystrophy in a child with
developmental delay: clinicopathological features and exclusion of the M1S1
gene. Eye (Lond) 2005;19(2):198-204. [CrossRef]
[PubMed]
19 Kawasaki S, Kinoshita S. Clinical and basic
aspects of gelatinous drop-like corneal dystrophy. Dev Ophthalmol 2011;48:97-115. [CrossRef]
[PubMed]
20 Magalhães Ode A, Rymer S, Marinho DR, Kwitko
S, Cardoso IH, Kliemann L. Optical coherence tomography image in gelatinous
drop-like corneal dystrophy: case report. Arq
Bras Oftalmol 2012;75(5):356-357. [CrossRef]
21 Kaji Y, Oshika T, Takazawa Y, Fukayama M,
Fujii N. Co-localisation of advanced glycation end products and D-beta-aspartic
acid-containing proteins in gelatinous drop-like corneal dystrophy. Br J Ophthalmol 2012;96(8):1127-1131. [CrossRef] [PubMed]
[PMC free article]
22 Edgington B, Goldstein MH. Anterior corneal dystrophy. In: Yanoff
M, Jay SD, Ophthalmology, 3rd ed. St Louis Mo: Mosby;2009:303-305. [CrossRef]
23 Javadi MA, Rezaei-Kanavi M, Javadi A,
Naghshgar N. Meesmann corneal dystrophy;a clinico-pathologic, ultrastructural
and confocal scan report. J Ophthalmic
Vis Res 2010;5(2):122-126. [PMC free article] [PubMed]
24 Corden LD, Swensson O, Swensson B, Smith FJ,
Rochels R, Uitto J, McLEAN WH. Molecular Genetics of MeesmannˇŻs Corneal
Dystrophy Ancestral and Novel Mutations in Keratin 12 (K12) and Complete
Sequence of the Human KRT12 Gene. Exp Eye
Res 2000;70(1):41-49. [CrossRef] [PubMed]
25 Kenyon KR, Hersh PS,
Starck T, et al. Corneal dysgeneses, dystrophies and degenerations. In: Tasman W.
DuaneˇŻs Clinical Ophthalmology. Philadelphia: Lippincott Williams & Wilkins
2004.
26 Szaflik JP, Oldak M, Maksym RB, Kamińska A,
Pollak A, Udziela M, Płoski R, Szaflik J. Genetics of Meesmann corneal
dystrophy a novel mutation in the keratin 3 gene in an asymptomatic family
suggests genotype phenotype correlation. Mol
Vis 2008;14:1713-1718. [PMC free article] [PubMed]
27 Ogasawara M, Matsumoto Y, Hayashi T, Dogru M,
Shimazaki J, Tsubota K, Tsuneoka H. KRT12 Mutations and in vivo confocal
microscopy in two japanese families with meesmann corneal dystrophy. Am J Ophthalmol 2014;157(1):93-102.e1. [CrossRef] [PubMed]
28 Spencer WH. Degenerations
and dystrophies. In: Spencer WH. Ophthalmic pathology, an atlas and textbook.
4th ed. Philadelphia: WB Saunders.
29 Reis W. Familiare,
fleckige Hornhautentartung. Dtsch Med
Wochenschr 1917;43:575.
30 B¨ącklers M. Über eine
weitere familiäre Hornhautdystrophie (Reis). Klin Monatsbl Augenheilkd
1949;114:386-397.
31 Joel Sugar, Wadia HP. Stromal corneal dystrophy. In: Yanoff M,
Duker JS. Ophthalmology. 3rd ed. Philadephia: Mosby Elsevier: 2009;306-311.
32 Piao MZ, Zhou XT, Wu LC, Chu RY. Arg555Gln
Mutation of TGFBI gene in geographical-type Reis-Bucklers corneal dystrophy in
a Chinese family. J Int Med Res 2012;40(3):1149-1155. [CrossRef]
33 Sayegh RR, Kouyoumjian PB, Vedula GG, Nottage
JM, Nirankari VS. Cocaine-assisted epithelial debridement for the treatment of
anterior basement membrane dystrophy. Cornea
2013;32(6):889-892. [CrossRef] [PubMed]
34 Hong JP, Kim TI, Chung JL, Huang D, Cho HS,
Kim EK. Analysis of deposit depth and morphology in granular corneal dystrophy
type 2 using fourier domain optical coherence tomography. Cornea 2011;30(7):729-738. [CrossRef] [PubMed]
35 Kim SW, Hong S, Kim T, Kim KS, Kim TI, Chung
WS, Kim EK. Characteristic features of granular deposit formation in granular
corneal dystrophy type 2. Cornea
2011;30(8):848-854. [CrossRef] [PubMed]
36 Choi SI, Kim BY, Dadakhujaev S, Oh JY, Kim TI,
Kim JY, Kim EK. Impaired autophagy and delayed autophagic clearance of
transforming growth factor beta-induced protein (TGFBI) in granular corneal
dystrophy type 2. Autophagy
2012;8(12):1782-1797. [CrossRef] [PubMed]
[PMC free article]
37 Francois J. Une nouvelle
dystrophie heredo-familiale de la cornee. Bull
Soc Belge Ophthalmol 1955(published in 1956);111:391-399.
38 Strachan IM. Cloudy central corneal dystrophy
of Francois. Five cases in the same family. Br
J Ophthalmol 1969;53(3):192-194. [CrossRef]
[PubMed] [PMC free article]
39 Karp CL, Scott IU, Green WR, Chang TS,
Culbertson WW. Central cloudy corneal dystrophy of Francois. A
clinicopathologic study. Arch Ophthalmol 1997;115(8):1058-1062. [CrossRef]
40 Belliveau MJ, Brownstein S, Agapitos P, Font
RL. Ultrastructural features of posterior crocodile shagreen of the cornea. Surv Ophthalmol 2009;54(5):569-575. [CrossRef] [PubMed]
41 Shearman AM, Hudson TJ, Andresen JM, Wu X,
Sohn RL, Haluska F, Housman DE, Weiss JS. The gene for schnyder's crystalline
corneal dystrophy maps to human chromosome 1p34.1-p36. Hum Mol Genet 1996;5(10):1667-1672. [CrossRef]
[PubMed]
42 Orr A, Dub¨¦ MP, Marcadier J, Jiang H, Federico
A, George S, Seamone C, Andrews D, Dubord P, Holland S, Provost S, Mongrain V,
Evans S, Higgins B, Bowman S, Guernsey D, Samuels M. Mutations in the UBIAD1
gene, encoding a potential prenyltransferase, are causal for Schnyder
crystalline corneal dystrophy. Plos One 2007;2(8):e685.
[CrossRef] [PubMed]
[PMC free article]
43 Yellore VS, Khan MA, Bourla N, Rayner SA, Chen
MC, Sonmez B, Momi RS, Sampat KM, Gorin MB, Aldave AJ. Identification of mutations
in UBIAD1 following exclusion of coding mutations in the chromosome 1p36 locus
for Schnyder crystalline corneal dystrophy. Mol
Vis 2007;13:1777-1782. [PubMed]
44 Weiss JS, Kruth HS, Kuivaniemi H, Tromp G,
Karkera J, Mahurkar S, Lisch W, Dupps WJ Jr, White PS, Winters RS, Kim C,
Rapuano CJ, Sutphin J, Reidy J, Hu FR, Lu da W, Ebenezer N, Nickerson ML.
Genetic analysis of 14 families with Schnyder crystalline corneal dystrophy
reveals clues to UBIAD1 protein function. Am
J Med Genet A 2008;146A(3):271-283. [CrossRef]
[PubMed]
45 Weiss JS, Kruth HS, Kuivaniemi H, Tromp G,
White PS, Winters RS, Lisch W, Henn W, Denninger E, Krause M, Wasson P,
Ebenezer N, Mahurkar S, Nickerson ML. Mutations in the UBIAD1 gene on
chromosome short arm 1, region 36, cause Schnyder crystalline corneal
dystrophy. Invest Ophthalmol Vis Sci 2007;48(11):5007-5012. [CrossRef]
[PubMed]
46 Ciancaglini M, Carpineto P, Doronzo E, Nubile
M, Zuppardi E, Mastropasqua L. Morphological evaluation of Schnyder's central
crystalline dystrophy by confocal microscopy before and after phototherapeutic
keratectomy. J Cataract Refract Surg
2001;27(11):1892-1895. [CrossRef]
47 Jing Y, Kumar PR, Zhu L, Edward DP, Tao S,
Wang L, Chuck R, Zhang C. Novel decorin mutation in a Chinese family with
congenital stromal corneal dystrophy.
Cornea 2014;33(3):288-293. [CrossRef] [PubMed]
48 Chen S, Sun M, Lozzo RV, Kao WW, Birk DE.
Intracellularly-retained decorin lacking the C-terminal ear repeat causes ER
stress: a cell-based etiological mechanism for congenital stromal corneal
dystrophy. Am J Pathol 2013;183(1):247-256.
[CrossRef] [PubMed]
[PMC free article]
49 Chen S, Sun M, Meng X, Iozzo RV, Kao WW, Birk
DE. Pathophysiological mechanisms of autosomal dominant congenital stromal
corneal dystrophy: C-terminal-truncated decorin results in abnormal matrix
assembly and altered expression of small leucine-rich proteoglycans. Am J Pathol 2011;179(5):2409-2419. [CrossRef] [PubMed] [PMC free article]
50 Francois J. A new hereditofamilial dystrophy
of the cornea. J Genet Hum 1956;5(3-4):189-196.
[PubMed]
51 Can E, Kan E, Akgun HI. Clinical features and
in-vivo confocal microscopic imaging of fleck corneal dystrophy. Semin Ophthalmol 2013;28(4):239-241. [CrossRef] [PubMed]
52 Purcell Jr JJ, Krachmer JH, Weingeist TA. Fleck corneal
dystrophy. Acta Ophthalmol 1977;95(3):440-444. [CrossRef]
53 Jiao X, Munier FL, Schorderet DF, Zografos L, Smith J, Rubin B,
Hejtmancik JF. Genetic linkage of Francois-Neetens fleck (mouchetee) corneal
dystrophy to chromosome 2q35. Hum Genet 2003;112(5-6):593-599. [PubMed]
54 Li S, Tiab L, Jiao X, Munier FL, Zografos L, Frueh BE, Sergeev
Y, Smith J, Rubin B, Meallet MA, Forster RK, Hejtmancik JF, Schorderet DF.
Mutations in PIP5K3 are associated with François-Neetens mouchet¨¦e fleck
corneal dystrophy. Am J Hum Genet 2005;77(1):54-63. [CrossRef] [PubMed] [PMC free article]
55 Vincent AL, Markie DM, De Karolyi B, Wheeldon CE, Patel DV,
Grupcheva CN, McGhee CN. Exclusion of known corneal dystrophy genes in an
autosomal dominant pedigree of a unique anterior membrane corneal dystrophy. Mol Vis 2009;15:1700-1708. [PMC free article]
[PubMed]
56 Nicholson DH, Green WR, Cross HE, Kenyon KR, Massof D. A
clinical and histopathological study of Francois-Neetens speckled corneal
dystrophy. Am J Ophthalmol 1977;83(4):554-560. [CrossRef]
57 Lakshminarayanan R, Chaurasia SS, Anandalakshmi V, Chai SM,
Murugan E, Vithana EN, Beuerman RW, Mehta JS. Clinical and Genetic Aspects of
the TGFBI-associated Corneal Dystrophies. The
Ocular Surface 2014;12(4):234-251.
[CrossRef] [PubMed]
58 Underhaug J, Koldsø H, Runager K, Nielsen JT, Sørensen CS,
Kristensen T, Otzen DE, Karring H, Malmendal A, Schiøtt B, Enghild JJ, Nielsen
NC. Mutation in transforming growth factor beta induced protein associated with
granular corneal dystrophy type 1 reduces the proteolytic susceptibility
through local structural stabilization. Biochim
Biophys Acta 2013;1834(12):2812-2822. [CrossRef] [PubMed] [PMC free article]
59 Skonier J, Neubauer M, Madisen L, Bennett K, Plowman GD,
Purchio AF. cDNA cloning and sequence analysis of beta ig-h3, a novel gene
induced in human adenocarcinoma cell line after treatment with transforming
growth factor-beta. DNA Cell Biol
1992;11(7):511-522. [CrossRef]
[PubMed]
60 Huerva V, Soldevila J, Matias-Guiu X. Recurrent amyloid
material in grafts used in patients with lattice corneal dystrophy 2
(MeretojaˇŻs syndrome). Med Hypothesis
Discov Innov Ophthalmol 2014;3(3):99-100. [PMC free article]
[PubMed]
61 Lisch W, Seitz B. Lattice Corneal dystrophy type 1: an
epithelial or stromal entity? Cornea 2014;33(10):1109-1112.
[CrossRef] [PubMed]
62 Jung JW, Kim SA, Kang EM, Kim TI, Cho HS, Kim EK. Lattice
corneal dystrophy type IIIA with hyaline component from a novel A620P mutation
and distinct surgical treatments. Cornea 2014;33(12):1324-1331. [CrossRef] [PubMed]
63 Akhtar S, Alkatan HM, Kirat O, Khan AA, Almubrad T. Collagen
fibrils and proteoglycans of macular dystrophy cornea: ultrastructure and 3d
transmission electron tomography. Microsc Microanal 2015;21(3):666-679. [CrossRef] [PubMed]
64 Kocluk Y, Yalniz-Akkaya Z, Burcu A, Ornek F. Corneal topography
analysis of stromal corneal dystrophies. Pak
J Med Sci 2015;31(1):116-120. [PMC free article]
[PubMed]
65 Dang X, Zhu Q, Wang L, Su H, Lin H, Zhou N, Liang T, Wang Z,
Huang S, Ren Q, Qi Y. Macular corneal dystrophy in a Chinese family related
with novel mutations of CHST6. Mol Vis 2009;15:700-705.
[PMC free article]
[PubMed]
66 Gruenauer-Kloevekorn C, Braeutigam S, Heinritz W, Froster UG,
Duncker GI. Macular corneal dystrophy: mutational spectrum in German patients,
novel mutations and therapeutic options. Graefes
Arch Clin Exp Ophthalmol 2008;246(10):1441-1447. [CrossRef] [PubMed]
67 Sultana A, Klintworth GK, Thonar EJ, Vemuganti GK, Kannabiran
C. Immunophenotypes of macular corneal dystrophy in India and correlation with
mutations in CHST6. Mol Vis 2009;15:319-325.
[PMC free article]
[PubMed]
68 Fernandez-Sasso D, Acosta JE, Malbran E. Punctiform and
polychromatic pre-Descemet's dominant corneal dystrophy. Br J Ophthalmol 1979;63(5):336-338. [CrossRef]
69 Friedmann NJ, Kaiser PK, Pineda R.
Pre-Descemet's Dystrophy. In: Friedmann NJ, Kaiser PK, Pineda R. The
Massachusetts Eye and Ear Infirmary Illustrated Manual of Ophthalmology, 3rd
ed. Saunders Elsevier:2009;223.
70 Kobayashi A, Ohkubo S, Tagawa S, Uchiyama K, Sugiyama K. In
vivo confocal microscopy in the patients with cornea farinata. Cornea 2003;22(6):578-581. [CrossRef]
71 Lanza M, Borrelli M, Benusiglio E, Rosa N. In vivo confocal
microscopy of an apparent deep stroma corneal dystrophy: a case report. Cases J 2009;2:9317. [CrossRef] [PubMed] [PMC free article]
72 Curran RE, Kenyon KR, Green WR. Pre-DescemetˇŻs membrane corneal
dystrophy. Am J Ophthalmol 1974;77(5):711-716.
[CrossRef]
73 Cockerham GC, Laver NV, Hidayat AA, McCoy DL. An
immunohistochemical analysis and comparison of posterior polymorphous dystrophy
with congenital hereditary endothelial dystrophy. Cornea 2002;21(8):787-791. [CrossRef]
74 Toma NM, Ebenezer ND, Inglehearn CF, Plant C, Ficker LA,
Bhattacharya SS. Linkage of congenital hereditary endothelial dystrophy to
chromosome 20. Hum Mol Genet 1995;4(12):2395-2398. [CrossRef]
75 Hand CK, Harmon DL, Kennedy SM, FitzSimon JS, Collum LM,
Parfrey NA. Localization of the gene for autosomal recessive congenital
hereditary endothelial dystrophy (CHED2) to chromosome 20 by homozygosity
mapping. Genomics 1999;61(1):1-4. [CrossRef] [PubMed]
76 Paliwal P, Sharma A, Tandon R, Sharma N, Titiyal JS, Sen S, Nag
TC, Vajpayee RB. Congenital hereditary endothelial dystrophy-mutation analysis
of SLC4A11 and genotype-phenotype correlation in a North Indian patient cohort. Mol Vis 2010;16:2955-2963. [PMC free article]
[PubMed]
77 Berger ST, McDermott ML, Aluri HKS. Corneal endothelium. In:
Yanoff M, Duker JS. Ophthalmology. 3rd ed. Mosby Elsevier: 2009;312-317. [CrossRef]
78 Cross HE, Maumenee A, Cantolino SJ. Inheritance of Fuchs'
endothelial dystrophy. Arch Ophthalmol 1971;85(3):268-272.
[CrossRef]
79 Krachmer JH, Purcell JJ Jr, Young CW, Bucher KD. Corneal
endothelial dystrophy: a study of 64 families. Arch Ophthalmol 1978;96(11):2036-2039. [CrossRef]
80 Zhang J, Patel DV. The pathophysiology of Fuchs' endothelial
dystrophy-a review of molecular and cellular insights. Exp Eye Res 2015;130:97-105. [CrossRef] [PubMed]
81 Engler C, Kelliher C, Spitze AR, Speck CL, Eberhart CG, Jun AS.
Unfolded protein response in Fuchs endothelial corneal dystrophy: a unifying
pathogenic pathway? Am J Ophthalmol
2010;149:194-202.e192. [CrossRef]
[PubMed] [PMC free article]
82 Hidayat AA, Cockerham GC. Epithelial metaplasia of the corneal
endothelium in Fuchs endothelial dystrophy. Cornea
2006;25(8):956-959. [CrossRef] [PubMed]
83 Naumann GO, Schlotzer-Schrehardt U. Keratopathy in
pseudoexfoliation syndrome as a cause of corneal endothelial decompensation: a
clinicopathologic study. Ophthalmology 2000;107(6):1111-1124. [CrossRef]
84 Davies Y, Fullwood NJ, Marcyniuk B, Bonshek R, Tullo A,
Nieduszynski IA. Keratan sulphate in the trabecular meshwork and cornea. Curr Eye Res 1997;16(7):677-686. [CrossRef]
85 Heon E, Geenberg A, Kopp KK, Rootman D, Vincent AL, Billingsley
G, Priston M, Dorval KM, Chow RL, McInnes RR, Heathcote G, Westall C, Sutphin
JE, Semina E, Bremner R, Stone EM. VSX1:a gene for posterior polymorphous
dystrophy and keratoconus. Hum Mol Genet 2002;11(9):1029-1036. [CrossRef]
86 Biswas S, Munier FL, Yardley J, Hart-Holden N, Perveen R,
Cousin P, Sutphin JE, Noble B, Batterbury M, Kielty C, Hackett A, Bonshek R,
Ridgway A, McLeod D, Sheffield VC, Stone EM, Schorderet DF, Black GC. Missense
mutations in COL8A2, the gene encoding the ¦Á2 chain of type VIII collagen,
cause two forms of corneal endothelial dystrophy. Hum Mol Genet 2001;10(21):2415-2423. [CrossRef]
87 Krafchak CM, Pawar H, Moroi SE, Sugar A, Lichter PR, Mackey DA,
Mian S, Nairus T, Elner V, Schteingart MT, Downs CA, Kijek TG, Johnson JM,
Trager EH, Rozsa FW, Mandal MN, Epstein MP, Vollrath D, Ayyagari R, Boehnke M,
Richards JE. Mutations in TCF8 cause posterior polymorphous corneal dystrophy
and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet 2005;77(5):694-708. [CrossRef] [PubMed] [PMC free article]
88 Liskova P, Tuft SJ, Gwilliam R, Ebenezer ND, Jirsova K,
Prescott Q, Martincova R, Pretorius M, Sinclair N, Boase DL, Jeffrey MJ,
Deloukas P, Hardcastle AJ, Filipec M, Bhattacharya SS. Novel mutations in the
ZEB1 gene identified in Czech and British patients with posterior polymorphous
corneal dystrophy. Hum Mutat 2007;28(6):638. [CrossRef] [PubMed] [PMC free article]
89 Aldave AJ, Yellore VS, YuF, Bourla N, Sonmez B, Salem AK,
Rayner SA, Sampat KM, Krafchak CM, Richards JE. Posterior polymorphous corneal
dystrophy is associated with TCF8 gene mutations and abdominal hernia. Am J Med Genet A 2007;143A(21):2549-2553. [CrossRef] [PubMed]
90 Henriquez AS, Kenyon KR, Dohlman CH, Boruchoff SA, Forstot SL,
Meyer RF, Hanninen LA. Morphologic characteristics of posterior polymorphous
dystrophy. A study of nine corneas and review of the literature. Surv Ophthalmol 1984;29(2):139-147. [CrossRef]
91 Brooks AM, Grant G, Gillies WE. Differentiation of posterior
polymorphous dystrophy from other posterior corneal opacities by specular
microscopy. Ophthalmology 1989;96(11):1639-1645. [CrossRef]
92 Hirst LW, Waring GO 3rd. Clinical specular microscopy of
posterior polymorphous endothelial dystrophy. Am J Ophthalmol 1983;95:143-155. [CrossRef]
93 Laganowski HC, Sherrard ES, Muir MG. The posterior corneal
surface in posterior polymorphous dystrophy: a specular microscopical study. Cornea 1991;10(3):224-232. [CrossRef]
94 Krachmer JH. Posterior polymorphous corneal dystrophy: a
disease characterized by epithelial-like endothelial cells which influence
management and prognosis. Trans Am
Ophthalmol Soc 1985;83:413-475.
[PMC free article]
[PubMed]
95 Weisenthal RW, Streeten B. Posterior
membrane dystrophies. In: Krachmer JH, Mannis MJ, Holland EJ, et al. Cornea and
external disease: clinical diagnosis and management. St Louis, MO:
Mosby;2005:1063-1090.
96 Sekundo W, Lee WR, Kirkness CM, Aitken DA, Fleck B. An
ultrastructural investigation of an early manifestation of the posterior
polymorphous dystrophy of the cornea. Ophthalmology
1994;101(8):1422-1431. [CrossRef]
97 Rodrigues MM, Newsome DA, Krachmer JH, Sun TT. Posterior
polymorphous dystrophy of the cornea: cell culture studies. Exp Eye Res 1981;33(5):535-544. [CrossRef]
98 Courtney DG, Atkinson SD, Allen EH, Moore JE, Walsh CP,
Pedrioli DM, MacEwen CJ, Pellegrini G, Maurizi E, Serafini C, Fantacci M, Liao
H, Irvine AD, McLean WH, Moore CB. siRNA silencing of the mutant keratin 12
allele in corneal limbal epithelial cells grown from patients with Meesmann's
epithelial corneal dystrophy. Invest
Ophthalmol Vis Sci 2014;55(5):3352-3560. [CrossRef] [PubMed]
[Top]