·Basic
Research··Current
Issue· ·Achieve· ·Search
Articles· ·Online Submission· ·About IJO· PMC
Comprehensive analysis
of genetic variations in strictly-defined Leber congenital amaurosis with whole-exome
sequencing in Chinese
Shi-Yuan
Wang, Qi Zhang, Xiang
Zhang, Pei-Quan Zhao
Department
of Ophthalmology,
Xinhua Hospital Affiliated to Shanghai Jiao Tong University
School of Medicine, Shanghai 200092, China
Correspondence to:
Pei-Quan Zhao. Department of Ophthalmology, Xinhua Hospital
Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 200092,
China. zhaopeiquan@126.com
Received: 2016-02-21
Accepted: 2016-03-11
Abstract
AIM: To make a comprehensive analysis
of the potential pathogenic genes related with Leber congenital
amaurosis (LCA) in Chinese.
METHODS: LCA subjects and their families
were retrospectively collected from 2013 to 2015. Firstly, whole-exome
sequencing was performed in patients who had underwent gene mutation screening
with nothing found, and then homozygous sites was selected, candidate sites were annotated, and pathogenic
analysis was conducted using softwares including Sorting Tolerant from Intolerant (SIFT), Polyphen-2, Mutation assessor,
Condel, and Functional Analysis through Hidden Markov Models (FATHMM). Furthermore, Gene Ontology
function and Kyoto
Encyclopedia of Genes and Genomes pathway enrichment analyses of pathogenic genes were performed followed by co-segregation
analysis using Fisher exact Test. Sanger sequencing was used to validate
single-nucleotide variations (SNVs). Expanded verification
was performed in the rest patients.
RESULTS: Totally 51 LCA
families with 53 patients and 24 family members were recruited. A total of 104 SNVs (66 LCA-related genes and
15 co-segregated genes) were submitted for expand verification. The frequencies
of homozygous mutation of KRT12 and CYP1A1 were simultaneously observed in 3
families. Enrichment analysis showed that the potential pathogenic genes were mainly enriched in functions related to cell
adhesion, biological adhesion, retinoid metabolic process, and eye
development biological adhesion.
Additionally, WFS1 and STAU2 had the highest homozygous
frequencies.
CONCLUSION: LCA is a highly
heterogeneous disease. Mutations in
KRT12, CYP1A1, WFS1, and STAU2 may be involved in the
development of LCA.
KEYWORDS:
Leber congenital amaurosis; whole-exome sequencing; targeted
next-generation sequencing
DOI:10.18240/ijo.2016.09.04
Citation:
Wang SY, Zhang Q, Zhang X, Zhao PQ. Comprehensive analysis of genetic
variations in strictly-defined Leber congenital amaurosis with whole-exome
sequencing in Chinese. Int J Ophthalmol 2016;9(9):1260-1264
INTRODUCTION
Leber
congenital amaurosis
(LCA)
is a rare inherited dystrophy of the retina, which is characterized by severe
loss of retinal and visual functions early in life with progressive
degeneration of the cellular structure of the retina[1]. LCA patients usually have poor visual function and
non-detectable or subnormal electroretinogram (ERG), and are
often accompanied by several complications such as nystagmus, photophobia, and
keratoconus[1]. Visual
acuity is rarely better than 20/400[2].
Presently, this disease affects approximately one in 80 000 of the
population[3]. About 20%
of children with LCA attend blind schools, accounting for about 5% of all
retinal dystrophies[4-6].
However, the molecular mechanisms underlying this disease are so complex to be
fully understood.
LCA
is a heterogeneous and autosomal recessive disease due to the abnormal
development of photoreceptor cells[1].
Broad expression variability is observed in patients with LCA, and the mechanisms of LCA disease
are involved with disruptions in phototransduction (AIPL1, GUCY2D), retinoid
cycle (RDH12, LRAT, RPE65),
photoreceptor development and structure (CRX,
CRB1), transport across the
photoreceptor connecting cilium (TULP1,
PRGRIP1, CEP290, LCA5), and other
ERG functions (IMPDH1, MERTK, RD3)[1].
Moreover, it has been well demonstrated that mutations in a single LCA gene can
lead to varied clinical phenotypes, and more than 60% LCA is caused by numerous
mutations in these genes[7].
However, these known genes share no specific regions that can be used as the
genetic markers for most LCA cases, and few clinical features are specific to
individual genetic abnormalities. Furthermore, genetic cause for
30%-50% patients suffering from LCA is still unclear, besides some other candidate genes have not
been identified.
Whole-exome
sequencing presents a broad
molecular background of disease, and can be used to distinguish new candidate
genes. Based on homozygosity mapping, whole-exome sequencing has been
successfully used to identify mutations in LCA-related genes[8-10].
In the present study, we aimed to make a comprehensive analysis of the
potential pathogenic genes related with LCA in Chinese. Briefly, whole-exome
sequencing was used to screen gene variants in parents and offsprings
of LCA pedigrees who had not ever been identified mutations in known LCA genes.
Then, the identified pathogenic variants were firstly verified via Sanger
sequencing, and then were further verified in another 41 patients. Specially,
this study only investigated the homozygous mutations, while was not involved
in compound heterozygous mutations and gene modifications. In addition, in
order to exclude patients with early onset retinitis pigmentosa or other
syndromic diseases that shared same phenotypes with LCA, clinical samples were
restricted to those who were < 1 year old and had
typical LCA phenotypes.
SUBJECTS AND METHODS
Subjects
This
was a retrospective
study conducted at
Xinhua Hospital Affiliated to
Shanghai Jiao Tong
University School of Medicine (Shanghai,
China). From May
2013 to November 2015, all subjects with LCA were collected.
They were performed fundus screening and ERG using RetCam¢ò
(Massie Research Laboratorles, Inc., USA).
All patients were recognized suitable if they met the following criteria: 1)
poor eyesight (without fixation, pendular
nystagmus, or the ability to follow light or subject) at birth or within one
year of age; 2) patients with extinguished ERG results. The following cases
were excluded: patients suffered from other congenital eye disease (retinopathy
of prematurity, congenital glaucoma, or familial
exudative vitreoretinopathy) or systemic hypoplasia
(hearing,
vestibular function, teeth, bone, muscle tension,
intelligence, liver, kidney, or blood sugar). The legal guardians of all
patients were provided with informed consent, and the protocol was approved by the
ethics committee of Xinhua Hospital.
Whole-exome Sequencing All
participants were subjected to whole-exome sequencing. Briefly, blood samples
were collected and the genome DNA was isolated using the Qiagen blood
genomic DNA extraction kit (Qiagen, Valencia, CA, USA) according to
the instructions. The DNA samples were ligated to paired-end adapters and
amplified by polymerase chain-reaction assay. Exome hybridization was performed
in all samples based on Ion Torrent platform and the whole exons sequencing was
performed by using Ion TorrentTM semiconductor sequencing system
(Life Technologies, USA).
Data Analysis Considering that LCA is usually an autosomal
recessive genetic disease, the homozygous sites were mainly selected as
reliable mutation sites. If a site was recessive homozygous in
the immediate family, it would be excluded. Then, the candidate sites were
annotated based on the single-nucleotide polymorphism database
(dbSNP) database, HapMap project,
1000 genomes project, and exome sequencing project using
SeattleSeq
SNP annotation and variant effect predictor in Ensemble
database. After excluding synonymous mutations and mutations
in intron area or untranslated region (UTR), the
mutations with biological functions were remained, including non-synonymous
mutation, frameshift mutation, splice site variants, and termination codon
mutations. Subsequently, pathogenic analysis of the mutations with biological
functions was carried out using softwares (SIFT, Polyphen-2,
Mutation assessor, Condel, FATHMM). Furthermore, Gene Ontology
(GO) function and Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathway enrichment analyses of pathogenic
genes were performed using database for annotation, visualization and
integration discovery (DAVID). P<0.05
was set as significant cut-off value for GO and KEGG pathway analysis. Moreover,
sight-related pathogenic genes were identified based on pathways which were
enriched by the known LCA-related genes, including pathways of eye development,
retinoidmetabolic process, sensory perception of light stimulus, visual
perception, vitamin A biosynthetic process, and photoreceptor cell maintenance.
Co-segregation Analysis The
disease information of pathogenic genes was obtained from DAVID, and the
expression information was extracted from BioGPS database which collected
expression information from 84 human tissues by using Affymetrix U133A arrays.
These pathogenic genes in both LCA patients and normal subjects were analyzed
using Fisher¡¯s exact test. The
non-empty hypothesis was that the frequency of single-nucleotide variations
(SNVs) was higher in LCA patients compared with normal subjects (P¡Ü0.05),
which was in order to verify whether genotype mutations were consistent with
the phenotypes of LCA or not.
Sanger Sequencing Sanger
sequencing was used to evaluate SNVs in each subject. All segments were
amplified through standard polymerase chain reaction (PCR),
and primers were designed using Primer Premier 5.0. The sequencing results were
analyzed through Chromas 2.2 software, and sequence alignment was performed to
exclude false positive sites.
Expand Verification SNVs
were further verified using targeted next-generation sequencing in the other 41
subjects. In brief, after preparation of sequencing template with Ion
PGM template OT2 200 kit (Life Technologies,
Gaithersburg, MD, USA), sequencing was performed using Ion proton 200
sequencing kit (Life Technologies, Carlsbad, USA) based on the Ion
PontonTM system.
RESULTS
Pedigrees
Totally,
ten families (family 1, 4, 7, 10, 11, 13, 14, 15, 24 and 37) were recruited in
the whole-exome sequencing, among which family 37 included two LCA patients
(patient identifiers: 37-3, 37-4). The clinical characteristics of these
patients were listed in Table 1. One member of family 4 (4-4, the aunt of 4-3)
had typical clinical symptoms of LCA (Figure 1) but the age of onset was
unclear. Specially, the previously known LCA-associated gene mutations were not
detected in 10 families in our hospital or other hospitals. Additionally, the
photographs of retina color for all the 11 LCA patients were exhibited in Figure
1.
Table
1 Clinical characteristics of the 11 LCA patients from 10 families
|
Patient ID |
Sex |
Visual
acuity (right/left) |
Nystagmus |
Fundus
change |
Vascular
changes |
Consan-guineous |
|
1-3 |
M |
LP/LP |
No |
Pigmentary
deposit |
Normal |
No |
|
4-3 |
F |
HM/HM |
No |
Leopard-like |
Attenuated |
Yes |
|
7-3 |
M |
HM/HM |
No |
Salt-pepper |
Attenuated |
No |
|
10-3 |
F |
LP/LP |
No |
Salt-pepper |
Attenuated |
No |
|
11-3 |
M |
LP/LP |
Yes |
Normal |
Normal |
No |
|
13-3 |
F |
FC/HM |
Yes |
Bone spicule |
Attenuated |
Yes |
|
14-3 |
F |
0.02/0.03 |
No |
Salt-pepper |
Attenuated |
No |
|
15-3 |
F |
0.1/0.1 |
No |
Nummular |
Attenuated |
No |
|
24-3 |
M |
LP/HM |
No |
Salt-pepper |
Normal |
Yes |
|
37-3 |
F |
0.03/0.02 |
No |
Fishnet
latticed |
Normal |
No |
|
37-4 |
M |
LP/HM |
No |
Fishnet
latticed |
Normal |
No |
LP: Light perception;
NLP: No light perception; FC: Finger counting; HM: Hand movement.
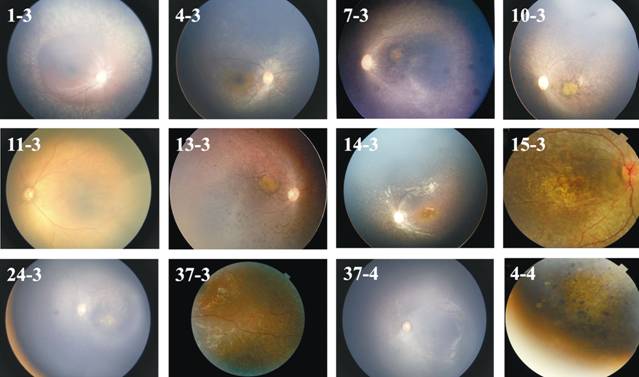
Figure 1 Color retinal
photographs of the 11 LCA subjects and one suspected member 4-4.
Functional and Pathway
Enrichment Analyses of Pathogenic Genes
GO functions and KEGG pathways enriched by the potential pathogenic
genes showed that the potential pathogenic genes were mainly enriched in functions related to cell
adhesion, biological adhesion, retinoid metabolic process, and eye
development biological adhesion. For instance, the pathogenic genes in
sample 13-3 were significantly enriched in the functions associated with cell
adhesion and biological adhesion. The pathogenic genes in sample 7-3 were
mainly enriched in pathways of viral myocarditis, olfactory transduction, and autoimmune
thyroid disease.
Leber Congenital Amaurosis-related Potential Pathogenic Gene Based
on the sight-related pathways enriched by previously known LCA-related genes,
68 LCA-related potential pathogenic genes
were identified, and their occurrence frequencies in the 11 samples were
calculated.
Among
these pathogenic genes, mutations in cytochrome
P450, family 1, subfamily A, polypeptide 1 (CYP1A1)
and eyes shut homologue (EYS) were
simultaneously found in four families, while EYS was not co-segregated. The mutation (amino acid Ile was
converted to Val) in CYP1A1 was
observed in three families (sample 1-3, 4-3 and 10-3).
Moreover, another two co-segregated genes were found in three families (sample
7-3, 13-3 and 24-3), including solute carrier family 22, member 16 (SLC22A16:
amino acid His was converted to Arg) and keratin 12 (KRT12:
amino
acid Pro was converted to Ser). SLC22A16
was not detected in retinal tissue.
Sanger Sequencing Fifty
percent of the potential LCA-related mutations (82) were randomly selected for
further validation within
the 11 samples. The results revealed that the accuracy of mutation detection
was 72%.
Expand Verification A total of 104
SNVs (66 sight-related genes
and 15 co-segregated genes) were submitted to expand
verification in another 41 samples. The frequencies of
homozygous mutation for KRT12 and CYP1A1 were respectively 0.333 and 0.118
in total 52 samples. In
addition, wolfram syndrome 1 (WFS1)
and staufen double-stranded RNA binding protein 2 (STAU2) had
the highest homozygous frequencies (0.941 and 0.922, respectively).
LCA
is usually considered as the most severe and earliest dystrophy of the retina,
which causes childhood blindness[11].
Half blindness in children is caused by genetic alterations, and some special
retinal appearance and longitudinal varies in visual function seem to be
gene-specific[12].
At present, Online Mendelian Inheritance In Man (OMIM) have recognized 18 types
of LCA. At least 22 LCA associated genes, such as RPE65, AIPL1, NMNAT1, and LCA5 have
been identified by linkage analysis with microsatellite markers or identity-by
descent mapping or the candidate gene method[13]. In the present study, a total of 104 novel
SNVs (66 sight-related genes and 15 co-segregated
genes) were identified. After co-segregation
analysis and verification, CYP1A1 and
KRT12 were simultaneously found in
three families, and WFS1 and
STAU2 had the highest homozygous frequencies.
Interestingly, the four genes in our study have not been discussed before,
which may be because they are specific to Chinese population.
Reportedly, CYP
genes
are involved in the process of organism response to environmental challenges,
and directly interfere with the embryo development[14].
Deleterious mutation of CYP1B1 causes
human primary congenital glaucoma via
disrupting the development of trabecular meshwork[15].
CYP1A1, an extrahepatic
phase ¦© metabolizing enzyme, is usually suppressed under physiologic
conditions and its expression is regulated by AHR (aryl hydrocarbon receptor)
signaling. The up-regulation and polymorphisms of CYP1A1 have been found to be associated with cell proliferation in
some cancers[16].
Moreover, CYP1A1 is up-regulated in
the retina by intraperitoneal injection of doxin, which finally induces
abnormal vascularization in the eye[17].
Dong et al[18]
demonstrated that mutants in CYP1A1
(Arg34Asp and Lys39Ile) could abolish the mitochondrial targeting signal.
However, to our knowledge, there is hardly any study about the role of CYP1A1 in LCA. In the present study, CYP1A1 mutations (Ile433Val, Ile434Val,
and Ile462Val)
were found in sample 1-3, 4-3 and 10-3, which had
a phenotype of thinner vascular. What¡¯s more, GO annotation suggested
that CYP1A1 was mainly involved in
functions related to retinoid metabolic process and eye development.
Therefore, we speculated that CYP1A1
mutation might be related to the process of LCA.
Keratin
proteins
are divided into type ¦© (KRT9-21) and type II (KRT1-8)
based on their sizes and isoelectric points, both of which are specifically and
predominantly expressed in corneal epithelial cells and function as obligate
heterodimers[19].
KRT12 is located on chromosome 17q12,
and several mutations in KRT12 can
cause Meesmann corneal epithelial dystrophy[20].
These mutations are located either in the highly conserved ¦Á-helix-initiation
motif of domain 1A or the ¦Á-helix-termination motif of domain 2B, which play
essential roles in the assembly of keratin
filament[19,21].
Besides, altered expression of keratin is also found in human retinal
pigment epithelial cells[22].
In the current study, a missense mutation (Pro15Ser)
of KRT12 was identified in LCA
samples. Additionally, function annotation suggested that KRT12 was involved in functions including sensory perception and
visual perception.
Therefore, mutation in KRT12 may be
associated with LCA.
In addition, our result
showed that WFS1 and STAU2 had the highest homozygous frequencies. WFS1 and STAU2 are expressed in the
central nervous system. WFS1 is
selectively expressed in neurons in different brain areas. Kawano et al[23] have suggested that the
mRNA and protein of WFS1
are detected in retina especially in amacrine and M¨¹ller cells, playing a
physiological role in normal visual system. Moreover, WFS1 mutation has an effect on the survival of retinal ganglion
cells and subsequently results in anterograde atrophy
of retinal axons[24]. STAU2, a double-strand mRNA binding protein, has been
reported to be associated with spinocerebellar ataxia, retinal
development, and eye morphogenesis[25].
Cockburn et al[26]
have found that ectopic expression of STAU2
increases eye size, and silencing of STAU2
results in a significantly reduced right/left eye diameter ratio in embryos. Combining with previous
studies, we speculated that WFS1
and STAU2 mutations might
have important effects on the development of LCA.
For
the treatment of LCA, gene therapy has been reported to be safe and effective.
In one form of LCA, patients bear a mutation in the RPE65 gene. In clinical trials, PRE65-targeted
therapy has been suggested to be a successful treatment for LCA through gene
augmentation therapy. Importantly, this therapeutic method can maintain the
vision function of LCA patients for more than three years[27].
Additionally, the therapeutic efficiencies of GUCY2D and RPGRIP1 on LCA
have been analyzed in LCA animal models[28-29].
In the present study, the newly detected genes might be involved in
vision-related pathways, therefore, it may
be worthy to carry out more in vivo and vitro studies with larger samples to confirm their
specific mechanism and further therapy efficiency.
ACKNOWLEDGEMENTS
Foundations:
Supported by National Natural Science Foundation of China
(No.81470642; No.81271045).
Conflicts
of Interest: Wang SY, None; Zhang Q, None;
Zhang X, None; Zhao PQ, None.
REFERENCES
1 den Hollander AI, Roepman
R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: Genes, proteins and
disease mechanisms. Prog Retin Eye Res
2008;27(4):391-419. [CrossRef] [PubMed]
2 Stone EM. Leber
congenital amaurosis-a model for efficient genetic testing of heterogeneous
disorders: Lxiv edward jackson memorial lecture. Am J Ophthalmol 2007;144(6):791-811. [CrossRef] [PubMed]
3 Allikmets R. Leber
congenital amaurosis: a genetic paradigm.
Ophthalmic Genet 2004;25(2):67-79. [CrossRef] [PubMed]
4 Schappert-Kimmijser J,
Henkes HE, Van Den Bosch J. Amaurosis congenita (leber). AMA Arch Ophthalmol 1959;61(2):211-218. [CrossRef]
5 Zulliger R, Naash MI,
Rajala RV, Molday RS, Azadi S. Impaired association of retinal degeneration-3
with guanylate cyclase-1 and guanylate cyclase activating protein-1 leads to
leber congenital amaurosis-1. J Biol Chem
2014;290(6):3488-3499. [CrossRef]
[PubMed] [PMC free article]
6 Weleber RG, Francis PJ,
Trzupek KM, Beattie C. Leber congenital amaurosis. GeneReviews®
[Internet]. Seattle (WA): University of Washington, Seattle;1993-2016.
7 den Hollander AI, Black
A, Bennett J, Cremers FP. Lighting a candle in the dark: Advances in genetics
and gene therapy of recessive retinal dystrophies. J Clin Invest 2010;120(9):3042-3053. [CrossRef] [PubMed] [PMC free article]
8 Wang X, Wang H, Cao M,
Li Z, Chen X, Patenia C, Gore A, Abboud EB, Al‐Rajhi AA, Lewis RA, Lupski JR, Mardon G, Zhang K,
Muzny D, Gibbs RA, Chen R. Whole‐exome sequencing identifies alms1, iqcb1, cnga3,
and myo7a mutations in patients with leber congenital amaurosis. Hum Mutat 2011;32(12):1450-1459. [CrossRef] [PubMed] [PMC free article]
9 Chiang P-W, Wang J,
Chen Y, et al. Exome sequencing identifies nmnat1 mutations as a cause of leber
congenital amaurosis. Nat Genet 2012;44(9):972-974. [CrossRef] [PubMed]
10 Wang X, Wang H, Sun V,
et al. Comprehensive molecular diagnosis of 179 leber congenital amaurosis and
juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet 2013;50(10):674-688. [CrossRef] [PubMed] [PMC free article]
11 Perrault I, Rozet JM,
Gerber S, Ghazi I, Leowski C, Ducroq D, Souied E, Dufier JL, Munnich A, Kaplan
J. Leber congenital amaurosis. Mol Genet
Metab 1999;68(2):200-208. [CrossRef] [PubMed]
12 Koenekoop RK, Lopez I,
Den Hollander AI, Allikmets R, Cremers FP. Genetic testing for retinal
dystrophies and dysfunctions: Benefits, dilemmas and solutions. Clin Experiment Ophthalmol
2007;35(5):473-485. [CrossRef] [PubMed]
13 Choudhary D, Jansson
I, Sarfarazi M, Schenkman J. Physiological significance and expression of p450s
in the developing eye. Drug Metab Rev 2006;38(1-2):337-352. [CrossRef] [PubMed]
14 Libby RT, Smith RS,
Savinova OV, Zabaleta A, Martin JE, Gonzalez FJ, John SW. Modification of
ocular defects in mouse developmental glaucoma models by tyrosinase. Science 2003;299(5612):1578-1581. [CrossRef] [PubMed]
15 Rodriguez M, Potter
DA. Cyp1a1 regulates breast cancer proliferation and survival. Mol Cancer Res 2013;11(7):780-792. [CrossRef] [PubMed] [PMC free article]
16 Zhan P, Wang Q, Qian
Q, Wei SZ, Yu LK. Cyp1a1 mspi and exon7 gene polymorphisms and lung cancer
risk: An updated meta-analysis and review. J
Exp Clin Cancer Res 2011;30:99.
[CrossRef] [PubMed] [PMC free article]
17 Takeuchi A, Takeuchi
M, Oikawa K, Sonoda KH, Usui Y, Okunuki Y, Takeda A, Oshima Y, Yoshida K, Usui
M. Effects of dioxin on vascular endothelial growth factor (vegf) production in
the retina associated with choroidal neovascularization. Invest Ophthalmol Vis Sci 2009;50(7):3410-3416. [CrossRef] [PubMed]
18 Dong H, Dalton TP,
Miller ML, Chen Y, Uno S, Shi Z, Shertzer HG, Bansal S, Avadhani NG, Nebert DW.
Knock-in mouse lines expressing either mitochondrial or microsomal cyp1a1:
differing responses to dietary benzo [a] pyrene as proof of principle. Mol Pharmacol 2009;75(3):555-567. [CrossRef] [PubMed] [PMC free article]
19 Corden LD, Swensson O,
Swensson B, Smith FJ, Rochels R, Uitto J, McLean WI. Molecular genetics of
meesmann's corneal dystrophy: Ancestral and novel mutations in keratin 12 (k12)
and complete sequence of the human krt12 gene. Exp Eye Res 2000;70(1):41-49. [CrossRef] [PubMed]
20 Ogasawara M, Matsumoto
Y, Hayashi T, Ohno K, Yamada H, Kawakita T, Dogru M, Shimazaki J, Tsubota K,
Tsuneoka H. Krt12 mutations and in vivo confocal microscopy in two japanese
families with meesmann corneal dystrophy. Am
J Ophthalmol 2014;157(1):93-102.e1. [CrossRef] [PubMed]
21 Nishida K, Honma Y,
Dota A, Kawasaki S, Adachi W, Nakamura T, Quantock AJ, Hosotani H, Yamamoto S,
Okada M, Shimonura Y, Kinoshita S. Isolation and chromosomal localization of a
cornea-specific human keratin 12 gene and detection of four mutations in
meesmann corneal epithelial dystrophy. Am
J Hum Genet 1997;61(6):1268-1275. [CrossRef] [PubMed] [PMC free article]
22 Hunt RC, Davis AA.
Altered expression of keratin and vimentin in human retinal pigment epithelial
cells in vivo and in vitro. J Cell
Physiol 1990;145(2):187-199. [CrossRef] [PubMed]
23 Kawano J, Tanizawa Y,
Shinoda K. Wolfram syndrome 1 (wfs1) gene expression in the normal mouse visual
system. J Comp Neurol 2008;510(1):1-23.
[CrossRef] [PubMed]
24 Yamamoto H, Hofmann S,
Hamasaki DI, Yamamoto H, Kreczmanski P, Schmitz C, Parel JM, Schmidt-Kastner R.
Wolfram syndrome 1 (wfs1) protein expression in retinal ganglion cells and
optic nerve glia of the cynomolgus monkey. Exp
Eye Res 2006;83(5):1303-1306. [CrossRef] [PubMed]
25 Raji B, Dansault A,
Leemput J, de la Houssaye G, Vieira V, Kobetz A, Arbogast L, Masson C, Menasche
M, Abitbol M. The RNA-binding protein musashi-1 is produced in the developing
and adult mouse eye. Mol Vis 2007;13:1412-1427.
[PubMed]
26 Cockburn DM, Charish
J, Tassew NG, Eubanks J, Bremner R, Macchi P, Monnier PP. The double-stranded
rna-binding protein staufen 2 regulates eye size. Mol Cell Neurosci 2012;51(3-4):101-111. [CrossRef] [PubMed]
27 Jacobson SG, Cideciyan
AV, Ratnakaram R, et al. Gene therapy for leber congenital amaurosis caused by
rpe65 mutations: Safety and efficacy in 15 children and adults followed up to 3
years. Arch Ophthalmol 2012;130(1):9-24.
[CrossRef] [PubMed] [PMC free article]
28 Williams ML, Coleman
JE, Haire SE, Aleman TS, Cideciyan AV, Sokal I, Palczewski K, Jacobson SG,
Semple-Rowland SL. Lentiviral expression of retinal guanylate cyclase-1
(retgc1) restores vision in an avian model of childhood blindness. PLoS Med 2006;3(6):e201. [CrossRef] [PubMed] [PMC free article]
29 Pawlyk BS, Smith AJ,
Buch PK, Adamian M, Hong DH, Sandberg MA, Ali RR, Li T. Gene replacement
therapy rescues photoreceptor degeneration in a murine model of leber
congenital amaurosis lacking rpgrip. Invest
Ophthalmol Vis Sci 2005;46(9):3039-3045. [CrossRef] [PubMed]
[Top]