
・Basic
Research・Current
Issue・ ・Achieve・ ・Search Articles・ ・Online Submission・ ・About IJO・ PMC
A Chinese family with
Axenfeld-Rieger syndrome: report of the clinical and genetic findings
Da-Peng Sun2, Yun-Hai Dai2, Xiao-Jing Pan2,
Tao Shan1, Dian-Qiang Wang2, Peng Chen1
1Qingdao University,
Qingdao 266071, Shandong Province, China
2State Key Laboratory
Cultivation Base, Shandong Provincial Key Laboratory of Ophthalmology, Shandong
Eye Institute, Shandong Academy of Medical Sciences, Qingdao 266071, Shandong
Province, China
Correspondence
to: Peng
Chen. Qingdao University, Qingdao 266071, Shandong Province, China.
chenpeng599205@126.com; Dian-Qiang Wang. State Key Laboratory Cultivation Base,
Shandong Provincial Key Laboratory of Ophthalmology, Shandong Eye Institute,
Shandong Academy of Medical Sciences, Qingdao 266071, Shandong Province, China.
qingcaodi2568@126.com
Received:
2016-09-22
Accepted: 2017-03-23
Abstract
AIM: To describe a
Chinese family affected by a severe form of Axenfeld-Rieger syndrome (ARS) and
characterize the molecular defect in PITX2 in the family.
METHODS: Patients
presented with typical ARS from a Chinese family were investigated. We
performed genome-wide linkage scan and exome sequencing to identify the
pathogenic mutations. Candidate mutations were verified for co-segregation in
the whole pedigree using Sanger sequencing. Real-time polymerase chain reaction
(RT-PCR) and Western blotting were performed to verify the expression of the
pathogenic gene.
RESULTS: Genome-wide
linkage and exome sequencing analyses showed PITX2 as the disease
candidate gene. A>G substitution at position -11 of 3’ss of exon 5
(IVS5-11A>G) that co-segregated with the disease phenotype was discovered in
the family. The PITX2 messenger ribonucleic acid and protein levels were
about 50% lower in patients with ARS than in unaffected family members in the
family.
CONCLUSION: Our findings
implicate the first intronic mutation of the PITX2 gene in the
pathogenesis of a severe form of ARS in a Chinese family. This study highlights
the importance of a systematic search for intronic mutation in ARS cases for
which no mutations in the exons of PITX2 have been found.
KEYWORDS: Axenfeld-Rieger syndrome;
exome sequencing; linkage analysis; PITX2; intronic mutation
DOI:10.18240/ijo.2017.06.04
Citation: Sun DP, Dai
YH, Pan XJ, Shan T, Wang DQ, Chen P. A Chinese family with Axenfeld-Rieger
syndrome: report of the clinical and genetic findings. Int J Ophthalmol 2017;10(6):847-853
Article Outline
INTRODUCTION
Axenfeld-Rieger
syndrome (ARS; OMIM 180500) is a rare hereditary multi-system disease with a
morbidity rate of 1:200 000. The genetic pattern of ARS is autosomal dominant
characterized by complete penetrance[1]. ARS has
several overlapping phenotypes, including Axenfeld anomalies, Rieger anomalies,
and Rieger syndrome[2].
The phenotype
of the anterior segment of ARS has significant heterogeneity, developmental
anomalies involving the cornea, iris, lens and angle[3].
The clinical phenotypes of the eye in ARS patients include iridogoniodysgenesis, posterior embryotoxon,
polycoria, corectopia, iris stromal hypoplasia, and iris strands bridging
the iridocorneal angle to the trabecular meshwork[4].
Anterior segmental dysplasia is the main cause of increased ocular pressure,
and about half of ARS patients will occur secondary glaucoma[5].
ARS-related
candidate pathogenic genes include PITX2 (paired-like homeodomain
transcription factor 2)[6-8] and
FOXC1 (forkhead box C1)[9-11].
Further study has found that chromosome site 13q14 is also associated with ARS.
The deletion of the 16q23-q24 region[12], the
deletion of the PAX6[13], and the missense
variant in the PRDM5[14] gene have been found in
ARS patients.
ARS in China
is relatively rare. Our purpose is to describe the clinical phenotype and
genetic characterization of a northeastern Chinese family affected by ARS.
SUBJECTS AND METHODS
Study Population
A Han Chinese
family with dominant ARS, and 200 matched, healthy controls were included. We
performed ophthalmic examinations for all participants, including vision,
ocular pressure measurement, corneal measurement, mirror microscopy, ultrasound
A/B scanning and ultrasound biomicroscopy, and used the slit lamp to take
pictures of the eyes. The study received written informed consent from all
participants and carried out in accordance with the Declaration of Helsinki.
Genome-wide
Linkage Analysis Using the genomic DNAs
from the ARS family, the whole genome linkage analysis was performed using
Illumina HumanOmniZhongHua-8 BeadChip. The specific steps were as follows, the
genomic DNA of each sample was amplified, fragmented, precipitated and
resuspended in a hybridization buffer. The denatured samples were hybridized on
Illumina HumanOmniZhongHua-8 BeadChip. Then the BeadChip oligonucleotides were
extended and detected on an Illumina Bead Array Reader by fluorescence imaging.
We used 872 261 SNPs to conduct linkage analysis after quality control
filtering. It was assumed that the disease allele frequency was 0.0001 with
dominant inheritance by a penetrance rate of 1. We performed multipoint
parametric linkage analysis in Merlin.
Whole Exome
Capture Total genomic DNA was
isolated from the venous blood of participants with the DNA isolation kit
(Tiangen, Beijing, China). Whole exome capture was performed by the Human All
Exon (50 Mb) target enrichment system (Agilent Technologies, Santa Clara, CA,
USA). Massive parallel sequencing was performed on the HiSeq 2500 Sequencing
System (Illumina Inc., San Diego, CA, USA). Consensus Coding Sequence Region
(CCDS) database provided the gene sequences in this array
(http://www.ncbi.nlm.nih.gov/projects/CCDS/). Berry Genomics Co., Ltd.
(Beijing, China) conducted the exome sequencing for the 3 individuals (III3,
III4, and IV2).
Variant
Analysis The sequencing data were
aligned with the human reference genome NCBI Build 36.3. The duplicated data
was delected, and SOAPaligner was used to compare the sequences of the 3
individuals. The SOAPsnp set was used to call the SNPs with the default
parameters. SNPs and short indels were provided as sequence variants. SNPs and
short indels were filtered against 1000 genome project
(http://www.1000genome.org/), HapMap 8 (http://hapmap.ncbi.nlm.nih.gov/)
database, the Single Nucleotide Polymorphism database (dbSNP,
http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi/), and YH database[15]. In view of familial ARS was rare, its pathogenic
mutations was believed to be rare too. Therefore, only less common and novel
variants were involved in the study. Less common variants meaned MAF (minor
allele frequency) was less than 0.05 in the 1000 Genomes Project.
Verification
of Variants Amplification primers for
candidate genes were designed and synthesized according to the genome sequence
of the human genome (hg18/build36.3). The target genes were amplified by
polymerase chain reaction (PCR) and the target gene sequences were obtained by
Sanger sequencing. In addition, the co-segregated mutantions were sequenced in
200 normal controls.
Ribonucleic Acid Extraction and Real-time Polymerase
Chain Reaction RNA isolation kit (Tiangen, Beijing, China) was used
to prepare total RNA from venous blood of all the family members. Total RNAs
were reverse transcribed into cDNA. SYBR Premix Ex Taq kit (Tiangen, Beijing,
China) was used in real-time PCR. The primer sequences for PITX2 were
5’-TTC ACA TCT GGC TCC ATG TT-3’ and 5’-GGG TTG CAT AGG CAG GTT AT-3’. The
glyceraldehyde-3-phosphate dehydrogenase gene was assessed with the primers
5’-ATG CTG GCG CTG AGT ACG T-3’ and 5’-AGC CCC AGC CTT CTC CAT-3’.
Western
Blotting and Antibodies The buffy coat of the
blood were harvested and lysed in RIPA buffer. The protein concentration
determined by the bicinchoninic acid (BCA) protein reagent (Beyotime). Equal
amounts protein samples were run on 10% SDS-PAGE gels, and subsequently
transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica,
MA, USA). The membranes were probed with primary antibodies. After incubated
with the secondary antibodies (HRP-conjugated goat anti-rabbit IgG), the signal
bands were visualized by enhanced chemiluminescence (ECL) Western blotting detection
reagent. GAPDH was served as the loading control.
RESULTS
Clinical
Phenotypes and Assessment We described a Han Chinese
family that had ARS with a dominant inheritance mode l (Figure 1). Five of 21
individuals in the four-generation family were identified to be affected with
ARS. Most eye phenotypes were bilateral and similar in severity. But the
severity degree was different among the patients. Ocular abnormalities of II2,
III1 and III4 included bilateral buphthalmos, corectopia, iris atrophy, and corneal
opacity (Figure 2A, 2C). Secondary glaucoma appeared during adolescence and
caused blindness in II2, III1 and III4. III3 had bilateral corectopia, iris
atrophy, and cataract (Figure 2B, Figure 3), and her IOP was 17 mm Hg in right
eye and 19 mm Hg in left eye. IV2 had bilateral corectopia and iris atrophy
(Figure 2D, Figure 4), and his IOP was 15 mm Hg in right eye and 16 mm Hg in
left eye.
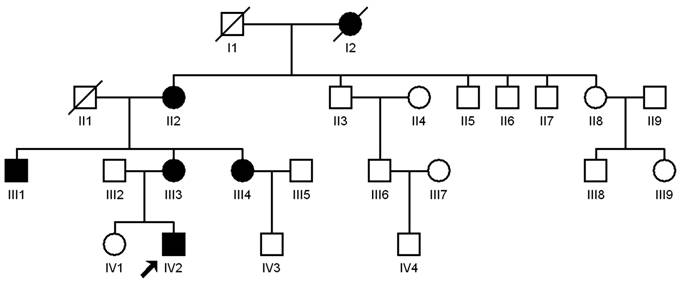
Figure 1 Pedigrees of the Chinese Axenfeld-Rieger
syndrome family The filled square represents a male
patient, and the filled circle indicates a female patient. An empty symbol
represents a normal individual. Forward slash represents the deceased person.

Figure 2 Clinical photographs of III1, III3, III4 and
IV2 affected with Axenfeld-Rieger syndrome The
patients in the family show similar clinical phenotypes, including a flattened midface, telecanthus, hypodontia or
microdontia.
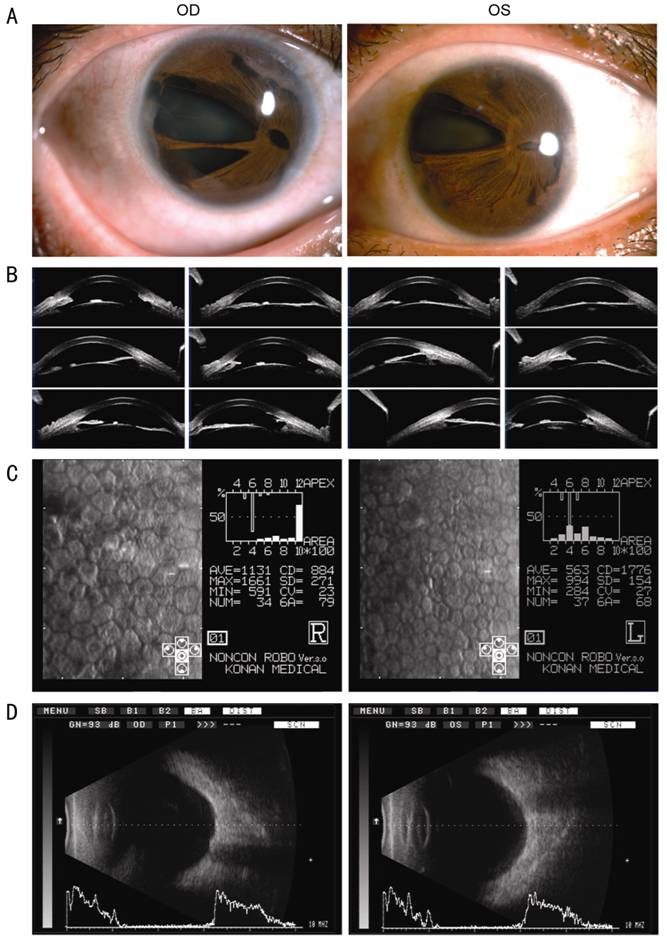
Figure 3 General pictutes (A), photographs of optical
coherence tomography (B), photographs of specular microscopy (C), and
ultrasound biomicroscopy (D) of patient III3 OD represents the right eye and OS represents left
eye.
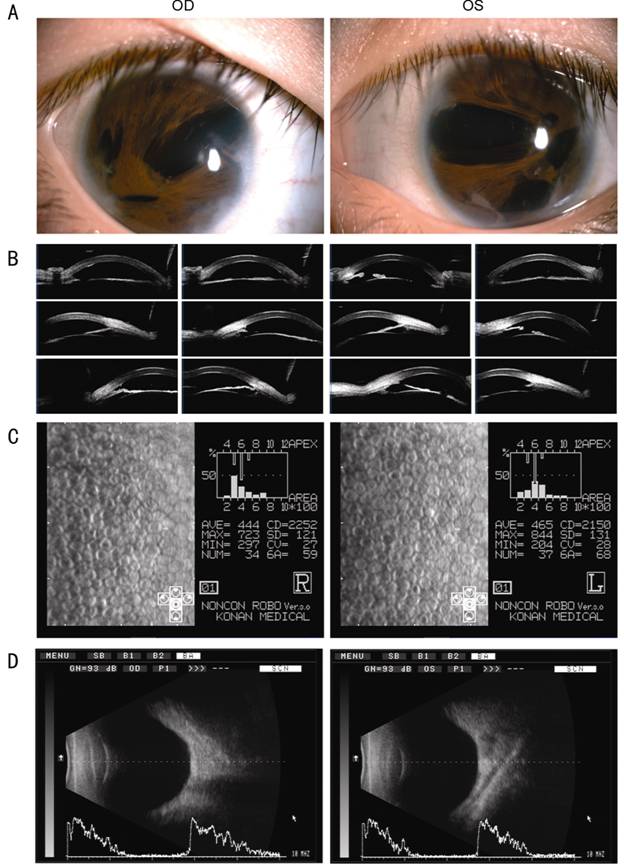
Figure 4 General pictutes (A), photographs of optical
coherence tomography (B), photographs of specular microscopy (C), and
ultrasound biomicroscopy (D) of patient IV2 OD
represents the right eye and OS represents left eye.
Five patients
in the family had similar typical facial features of ARS, such as a broad nasal
bridge, a broad forehead, flattening of the midface, a thin upper lip with a
long philtrum, a protruding lower lip, hypodontia, and microdontia (Figure
2E-2H). The five patients had no hearing impairment and cardiovascular defects.
Causal
Regions Identified by Genome-wide Linkage
The
study identified eight susceptibility loci, rs1107550-rs4660192 (Chr1:
32048052-Chr1: 41842350; 9.8 Mb), kgp1419269-kgp12301015 (Chr4: 83673890-Chr4:
133385086; 49.7 Mb), rs877741-rs11744690 (Chr5: 148196737-Chr5: 158687153; 10.5
Mb genomic region), rs10904561-rs583227 (Chr10: 135656-Chr10: 13166875; 13 Mb),
kgp5435398-rs1711782 (Chr13: 46707064-Chr13: 60101738; 13.4 Mb), rs332233-rs6495314
(Chr15: 64367745-Chr15: 78960529; 14.6 Mb), rs8044984-rs2037174 (Chr16:
23361853-Chr16: 27126459; 3.8 Mb), and rs12982096-rs3760961 (Chr19:
288738-Chr19: 3013374; 2.7 Mb), with a LOD score of 1.505.
Associated
PITX2 Mutation in the Axenfeld-Rieger Syndrome Family We focused on finding the pathogenic
genes in these candidate regions by exome sequencing (III3, III4, and IV2).
Through the exon subgroup sequencing, each sample produced an average of 7.41
billion base sequencing data. BWA was used to compare the data with the hg19
human reference genome[16]. The average
sequencing depth of each sample was 67.54. An average of 86.74% of the exon
sequences were sequenced at least 10X. Single nucleotide variants (SNV) and
indels were filtered by GATK Unified Genotyper[17].
The filtered
SNV and indels were annotated. Then they were filtered against 1000 genome
project, HapMap 8 database, the SNP database, and YH database. In view of the
synonymous variants were unlikely to be pathogenic. We screened out non-synonymous
mutations (nonsense, missense and read-through), encoding indels, and variants
of splice donor and acceptor sites. The filtered data was shown in Table 1.
Table 1 Single nucleotide variants (SNV) and indels indentified in III3,
III4, and IV2 through exome resequencing
|
Filter1 |
Genetic variants |
|||||
|
SNV: exon |
SNV: splicing site |
SNV: intron |
SNV: UTR |
Indel |
Total |
|
|
III3 |
925 |
31 |
738 |
150 |
2227 |
4071 |
|
III4 |
1069 |
50 |
823 |
163 |
2308 |
4413 |
|
IV2 |
909 |
24 |
658 |
141 |
2332 |
4064 |
|
Heterozygous shared by five
affected individuals non-synonymous in the linkage regions |
12 |
0 |
3 |
2 |
29 |
46 |
1Not in 1000 Genomes
Project (MAF≥0.05), the dbSNP, HapMap 8, YH database.
Twelve
non-synonymous SNVs, 3 introns, 29 indels and 2 UTRs were screened out by
recommended filtering criteria. Only one SNV (chr4:110618699) was located in
the linkage region and was co-segregated in the ARS pedigree. It lied in the
upstream of the 3’ss of exon 5 of PITX2 (NC_00000.12), an A>G change
(IVS5-11A>G). The mutation didn’t in the 200 control individuals (Figure 5).
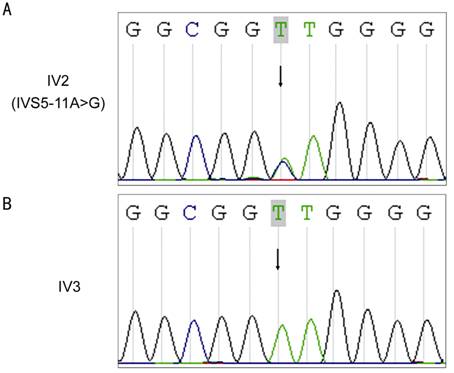
Figure 5 The sequence of the PITX2
gene chr4: 110618699 in the ARS family A represents patient IV2; B represents
the normal individual IV3. The arrow indicates chr4: 110618699.
PITX2 Expression Analysis We investigated the effect of the
detected mutation on PITX2 expression in the ARS family. Compared with
normal individuals in the pedigrees, the expression level of PITX2 mRNA
was reduced by about 46% in patients (P<0.01; Figure 6). And the
expression level of PITX2 protein in patients decreased by about 60% (P<0.01;
Figure 6).
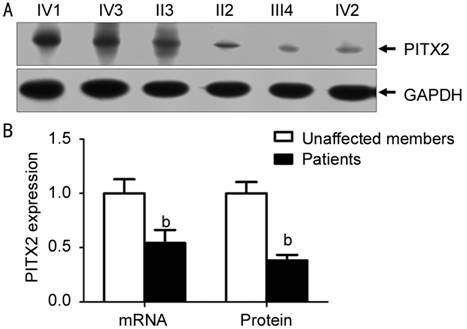
Figure 6 The effect of the
detected mutation on PITX2 expression in the ARS family A: The expression level of PITX2
protein in patients and normal individuals deteceted by Western blotting; B: PITX2
protein and mRNA expression levels were applied to the mean±SD. bP<0.01
compared with normal individuals in the pedigrees.
DISCUSSION
ARS patients
usually have a variety of systemic abnormalities, including facial and dental
deformities. The main abnormalities of the face include prominent forehead, a
broad and flat nasal bridge, maxillary hypoplasia with flattening of the
midface, and telecanthus. Dental abnormalities include hypodontia or
microdontia. In the abdomen, abnormal skin evolution caused by the accumulation
of skin around the navel. In addition, in some ARS patients, there are various
clinical phenotypes, such as growth retardation, pituitary abnormalities, anal
stenosis, and hypospadias unspecified. However, exceptions to the above
mentioned phenotypic abnormalities are not considered to be the classical
features of ARS[18].
High
permeability in eye morphogenesis of ARS patients are associated with the
development of glaucoma. About 50% of ARS patients develop secondary glaucoma.
Glaucoma may occur in childhood, but it is more common in adolescence or
adulthood. In some cases it may occur even after middle age. Treatment of
secondary glaucoma is usually tricky and can lead to severe disc damage and
visual field defects. In our study, secondary glaucoma appeared during adolescence
and caused blindness in II2, III1 and III4. Bilateral eye became buphthalmos
and suffered devastating damage. But III3 didn’t have secondary glaucoma at the
age of 40.
PITX2 belongs to the family of
bicoid-like homeobox transcription factor. The members of homeobox gene family
play important roles in the development of the individual, especially in the
pattern formation and cell fate decisions. PITX2 is expressed in neural
crest cells and is essential for the normal development of optic stalk and formation
of the anterior segment structures[19]. There are
at least four different PITX2 transcription isforms, PITX2a, PITX2b,
PITX2c and PITX2d. They have different biological properties respectively[20]. PITX2a, PITX2b and PITX2c contain the same homologous
domain and COOH-terminus, but there are differences in structure at the
NH2-terminus. PITX2d is truncated, resulting in a nonfunctional homology domain[20]. Although the effect and specific mechanism of PITX2
in the pathogenesis of ARS are still unclear, the lack of normal expression of
PITX2 protein is considered to be one of the main molecular mechanisms of ARS
development[21]. Up to now, several intron
mutations have been reported to cause ARS[22-23], but the vast majority of ARS-induced mutations are
mostly located in the COOH terminal domain and homologous domain. Through the
integration of Wnt and signal retinoic acid, it is found that PITX2 is the key
molecule in the anterior segment model[24].
Mutations within the HD domain can hinder the ability of the protein to bind to
the homologous DNA target sequence, leading to abnormal regulation of the
target genes.
Our study
implicated the intronic mutation of the PITX2 gene associated with the
pathogenesis of ARS in China. It revealed that this intron mutation described
in the previous literature may affect the expression of PITX2 protein and
trigger the pathogenesis of ARS. An A>G change 11 nt upstream of the 3’ss of
exon 5 (IVS5-11A>G) of PITX2 was co-segregated with the disease
phenotype in the ARS family[25]. The
polypyrimidine was located between the branch point and the splice point.
Polypyrimidines played an important role in the strength of 3’ss. The
disruption of purine resulted in a 3’ss quality reduction. The strength of the
3’ss may not be altered by the IVS5-11A>G mutation, but a new “AG”
dinucleotide was produced. It is assumed that the new “AG” could compete with
the original one 11 nt downstream[26].
In conclusion,
our study reported for the first time a mutation in the intron region that triggered
the pathogenesis of a Chinese ARS family. The analysis of the expression level
of PITX2 further confirmed the possibility of development of ARS induced
by PITX2 haploid deficiency. At the same time, we summarized the
variable phenotype in five patients in the ARS family and expanded the clinical
phenotype profile of ARS in a different racial background.
ACKNOWLEDGEMENTS
We would like
to express our gratitude to the participating family. For the data set used to
filter variants, we thank the Single Nucleotide Polymorphism database, 1000
Genome Project, HapMap 8 database, and YH database.
Foundations: Supported by China
Postdoctoral Science Foundation Funded Project (No.2017M612211); the National
Natural Science Foundation of China (No.81300742; No.81600721); the Shandong
Province Medical and Health Technology Development Project (No.2016WS0265); the
Science and Technology Plan of Qingdao (No.15-9-1-35-jch).
Conflicts
of Interest: Sun DP, None; Dai YH, None; Pan XJ, None; Shan
T, None; Wang DQ, None; Chen P, None.
REFERENCES
1 Hjalt TA, Semina EV. Current molecular understanding of
Axenfeld-Rieger syndrome. <ii>Expert Rev Mol Med</ii>
2005;7(25):1-17. [PubMed]
2 Alward WL. Axenfeld-Rieger syndrome in the age of molecular
genetics. <ii>Am J Ophthalmol</ii> 2000;130(1):107-115.[CrossRef]
3 Ito YA, Walter MA. Genomics and anterior segment dysgenesis: a
review. <ii>Clin Exp Ophthalmol</ii> 2014;42(1):13-24. [PubMed]
4 Tumer Z, Bach-Holm D. Axenfeld-Rieger syndrome and spectrum of
PITX2 and FOXC1 mutations. <ii>Eur J Hum Genet</ii>
2009;17(12):1527-1539. [PMC free article] [PubMed]
5 Idrees F, Vaideanu D, Fraser SG, Sowden JC, Khaw PT. A review of
anterior segment dysgeneses. <ii>Surv Ophthalmol</ii> 2006;51(3):213-231.
[PubMed]
6 Seifi M, Footz T, Taylor SA, Elhady GM, Abdalla EM, Walter MA.
Novel PITX2 gene mutations in patients with Axenfeld-Rieger syndrome.
<ii>Acta Ophthalmol</ii> 2016;94(7):e571-e579. [PubMed]
7 Yin HF, Fang XY, Jin CF, Yin JF, Li JY, Zhao SJ, Miao Q, Song
FW. Identification of a novel frameshift mutation in PITX2 gene in a Chinese
family with Axenfeld-Rieger syndrome. <ii>J Zhejiang Univ Sci
B</ii> 2014;15(1):43-50. [PMC free article] [PubMed]
8 Reis LM, Tyler RC, Volkmann Kloss BA, <ii>et
al</ii>. PITX2 and FOXC1 spectrum of mutations in ocular syndromes.
<ii>Eur J Hum Genet</ii> 2012; 20(12):1224-1233. [PMC free article] [PubMed]
9 Micheal S, Siddiqui SN, Zafar SN, Villanueva-Mendoza C,
Cortés-González V, Khan MI, den Hollander AI. A novel homozygous mutation in
FOXC1 causes Axenfeld Rieger Syndrome with congenital glaucoma. <ii>PLoS
One</ii> 2016;11(7):e0160016. [PMC free article] [PubMed]
10 Yang HJ, Lee YK, Joo CK, Moon JI, Mok JW, Park MH. A family
with Axenfeld-Rieger Syndrome: report of the clinical and genetic findings.
<ii>Korean J Ophthalmol</ii> 2015;29(4):249-255. [PMC free article] [PubMed]
11 Du RF, Huang H, Fan LL, Li XP, Xia K, Xiang R. A novel mutation
of FOXC1 (R127L) in an Axenfeld-Rieger syndrome family with glaucoma and
multiple congenital heart diseases. <ii>Ophthalmic Genet</ii>
2016;37(1): 111-115. [PubMed]
12 Werner W, Kraft S, Callen DF, Bartsch O, Hinkel GK. A small
deletion of 16q23.1->16q24.2 [del(16)(q23.1q24.2).ish
del(16)(q23.1q24.2)(D16S395+, D16S348-, P5432+)] in a boy with iris coloboma
and minor anomalies. <ii>Am J Med Genet</ii> 1997;70(4):371-376. [CrossRef]
13 Riise R, Storhaug K, Brondum-Nielsen K. Rieger syndrome is
associated with PAX6 deletion. <ii>Acta Ophthalmol Scand</ii>
2001;79(2): 201-203. [CrossRef]
14 Micheal S, Siddiqui SN, Zafar SN, Venselaar H, Qamar R, Khan
MI, den Hollander AI. Whole exome sequencing identifies a heterozygous missense
variant in the PRDM5 gene in a family with Axenfeld-Rieger syndrome.
<ii>Neurogenetics</ii> 2016;17(1):17-23. [PMC free article] [PubMed]
15 Li G, Ma L, Song C, <ii>et al</ii>. The YH
database: the first Asian diploid genome database. <ii>Nucleic Acids
Res</ii> 2009;37(Database issue): D1025-D1028. [PMC free article] [PubMed]
16 Li H, Durbin R. Fast and accurate short read alignment with
Burrows-Wheeler transform. <ii>Bioinformatics</ii>
2009;25(14):1754-1760. [PMC free article] [PubMed]
17 McKenna A, Hanna M, Banks E, <ii>et al</ii>. The
Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA
sequencing data. <ii>Genome Res</ii> 2010;20(9):1297-1303. [PMC free article] [PubMed]
18 Burkitt Wright EMM, Spencer HL, Daly SB, <ii>et
al</ii>. Mutations in PRDM5 in brittle cornea syndrome identify a pathway
regulating extracellular matrix development and maintenance. <ii>Am J Hum
Genet</ii> 2011;88(6):767-777. [PMC free article] [PubMed]
19 Evans AL, Gage PJ. Expression of the homeobox gene Pitx2 in
neural crest is required for optic stalk and ocular anterior segment
development. <ii>Hum Mol Genet</ii> 2005;14(22):3347-3359. [PubMed]
20 Cox CJ, Espinoza HM, McWilliams B, Chappell K, Morton L, Hjalt
TA, Semina EV, Amendt BA. Differential regulation of gene expression by PITX2
isoforms. <ii>J Biol Chem</ii> 2002;277(28):25001-25010. [PubMed]
21 Lines MA, Kozlowski K, Kulak SC, <ii>et al</ii>.
Characterization and prevalence of PITX2 microdeletions and mutations in
Axenfeld-Rieger malformations. <ii>Invest Ophthalmol Vis Sci</ii>
2004;45(3):828-833. [CrossRef]
22 Maciolek NL, Alward WL, Murray JC, Semina EV, McNally MT.
Analysis of RNA splicing defects in PITX2 mutants supports a gene dosage model
of Axenfeld-Rieger syndrome. <ii>BMC Med Genet</ii> 2006;7:59. [PMC free article] [PubMed]
23 de la Houssaye G, Bieche I, Roche O, <ii>et
al</ii>. Identification of the first intragenic deletion of the PITX2
gene causing an Axenfeld-Rieger Syndrome: case report. <ii>BMC Med
Genet</ii> 2006;7:82. [PMC free article] [PubMed]
24 Gage PJ, Qian M, Wu D, Rosenberg KI. The canonical Wnt
signaling antagonist DKK2 is an essential effector of PITX2 function during
normal eye development. <ii>Dev Biol</ii> 2008;317(1):310-324. [PMC free article] [PubMed]
25 Borges AS, Susanna R Jr, Carani JC, Betinjane AJ, Alward WL,
Stone EM, Sheffield VC, Nishimura DY. Genetic analysis of PITX2 and FOXC1 in
Rieger Syndrome patients from Brazil. <ii>J Glaucoma</ii>
2002;11(1):51-56. [CrossRef]
26 Zeniou M, Gattoni R, Hanauer A, Stevenin J. Delineation of the
mechanisms of aberrant splicing caused by two unusual intronic mutations in the
RSK2 gene involved in Coffin-Lowry syndrome. <ii>Nucleic Acids
Res</ii> 2004;32(3):1214-1223. [PMC free article] [PubMed]