
・Basic Research・ Current
Issue IF in JCR CiteScore ・Submission・ In Press Recent Accepted PMC RSS
Citation: Gong HM, Wang J, Xu J, Zhou ZY, Li JW, Chen SF. Identification of rare
paired box 3 variant in strabismus by whole exome sequencing. Int J
Ophthalmol 2017;10(8):1223-1228
Identification of rare paired box 3 variant in
strabismus by whole exome sequencing
Hui-Min Gong1, Jing Wang2,
Jing Xu3, Zhan-Yu Zhou1, Jing-Wen Li1,
Shu-Fang Chen4
1Ophthalmologic Center, Qingdao Municipal Hospital, the Affiliated
Municipal Hospital of Qingdao University, Qingdao 266000, Shandong Province,
China
2Department of Ophthalmology, Dezhou People’s Hospital, Dezhou
253000, Shandong Province, China
3Department of Ophthalmology, Weifang People's Hospital, Weifang
261041, Shandong Province, China
4Department of Medical Equipment, Weifang People's Hospital,
Weifang 261041, Shandong Province, China
Correspondence
to: Shu-Fang Chen. Department of Medical Equipment, Weifang People's
Hospital, No.151 Kuiwen District, Guangwen Street, Weifang 261041, Shandong
Province, China. 13869662816@163.com
Received:
2017-02-23
Accepted: 2017-04-24
Abstract
AIM: To
identify the potentially pathogenic gene variants that contributes to the
etiology of strabismus.
METHODS: A
Chinese pedigree with strabismus was collected and the exomes of two affected
individuals were sequenced using the next-generation sequencing technology. The
resulting variants from exome sequencing were filtered by subsequent bioinformatics
methods and the candidate mutation was verified as heterozygous in the affected
proposita and her mother by sanger sequencing.
RESULTS:
Whole exome sequencing and filtering identified a nonsynonymous mutation
c.434G-T transition in paired box 3 (PAX3) in the two affected individuals,
which were predicted to be deleterious by more than 4 bioinformatics programs.
This altered amino acid residue was located in the conserved PAX domain of
PAX3. This gene encodes a member of the PAX family of transcription factors,
which play critical roles during fetal development. Mutations in PAX3 were
associated with Waardenburg syndrome with strabismus.
CONCLUSION:
Our results report that the c.434G-T mutation (p.R145L) in PAX3 may contribute
to strabismus, expanding our understanding of the causally relevant genes for
this disorder.
KEWORDS:
strabismus; whole exome sequencing; paired box 3
DOI:10.18240/ijo.2017.08.06
Citation: Gong HM, Wang J, Xu J, Zhou ZY, Li JW, Chen SF. Identification of rare
paired box 3 variant in strabismus by whole exome sequencing. Int J
Ophthalmol 2017;10(8):1223-1228
INTRODUCTION
Strabismus
is a common ocular disorder which is characterized by the misalignment of the
eyes[1-4]. Strabismus is often
associated with amblyopia of children, which can cause visual disturbance[1]. It is reported that the prevalence of strabismus is
2.4% in Hispanic/Latinos, 2%-4% in Caucasians, 2.5% in African-Americans, and
1% in East-Asians[5-8]. Additionally,
the incidence of specific types of strabismus also shows differences in
different racial groups, in which Asian strabismus are exotropia, suggesting
the relevance of genetic factors[9-11].
Summing data from the medical literature show that the etiology of strabismus
has a genetic component because the familial clustering of strabismus has been
recognized[12-13].
Three
inheritance patterns including dominant, recessive and sex-linked have been associated
with nonsyndromic strabismus in family studies[14-15]. Parikh et al[16] found
that a family of nonsyndromic strabismus conformed to the recessive inheritance
model, and they identified susceptibility locus 7p22.1 with a multipoint LOD
score of 4.51. However, linkage to 7p in 6 other families was not observed. In
addition, 7p22.1 of dominant inheritance model, 16p13.12-p12.3 of recessive
inheritance model and 4q28.3 dominant inheritance model has been selected as
comitant strabismus associated locus[17-19].
Considering the genetic heterogeneity among families, the identity of the
relevant candidate genes remains a challenge. Further work should be conducted
to identify more causally relevant genes, improving the understanding of this
disorder.
After
the exciting finding that exomes sequencing was first developed in 2009[20], exome sequencing was widely used to locate causative
genes in rare Mendelian diseases or complex diseases with high sensitivity and
specificity[21-25]. Advances
in genetic methodology may provide insight into the genetic basis for inherited
strabismus. In the current study, we used whole exome sequencing to identify
the causative gene for the two affected individuals in a Chinese strabismus
family. Several evidences supported the causal role of paired box 3 (PAX3) in
strabismus susceptibility.
SUBJECTS AND METHODS
Subjects For the
purpose of this study, a three-generation Chinese strabismus family with two
affected individuals was recruited (Sample II:2 and III:1) (Figure 1A). The
proposita was a 7-year-old girl from Shandong Province who presented with
intermittent exotropia of unknown etiology, leading to amblyopia (uniocular
visual neglect). Once she was tired, one of the eyes will turn outwards
intermittently when looking into the distance. The amount of tropia was -50△
(near) and -50△ (far) and the unaided visual acuity was 1.0 in the right eye
and 1.0 in the left eye. Moreover, her mother was 35-year-old and also
presented with intermittent exotropia with similar phenotypes. The amount of
tropia was -40△ (near) and -40△ (far). Her best-corrected visual acuity was OD
1.0 and OS 1.0. Given the high suspicion for a congenital strabismus family,
the two affected individuals were enrolled for the exome sequencing screen.
Peripheral blood samples were collected in EDTA tubes from the participants for
DNA extraction. The written informed consent was then obtained from study
subjects or guardian before the study. Our study was approved by the Ethics
Committee of the Affiliated Hospital of Qingdao University (2015-012).
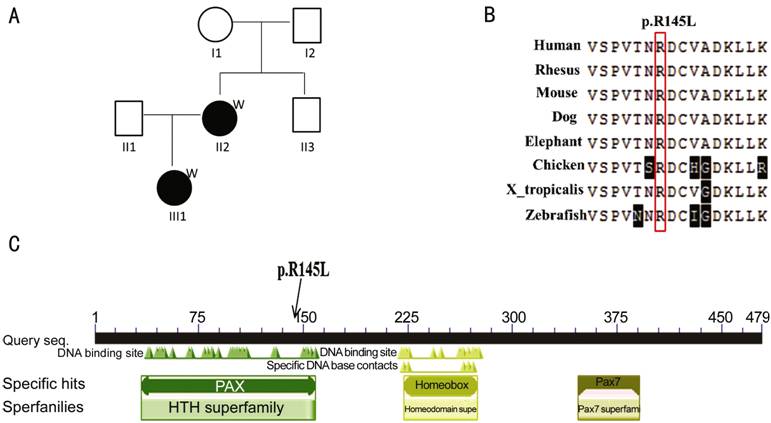
Figure
1 The analysis of PAX3 in strabismus A: Pedigree for the Chinese family with
strabismus, individuals II:2 and III:1 underwent exome sequencing; B: Affected
amino acid residue was highly conserved across species; C: Conserved domains in
PAX3. The mutation c.434G-T (p.145R-L) was located in the conserved PAX domain
of PAX3.
Exome
Capture Analysis Genomic DNA
was extracted from the blood samples obtained from the available patients
according to the standard procedures. The 2 μg of genomic DNA was fragmented
with about 200 bp, then ligated with adapters and amplified by
ligation-mediated polymerase chain reaction (PCR). The qualified genomic DNA
was used for exome capture and high-throughput sequencing. Agilent SureSelect
Human All Exon 50 Mb Exon Kit was used to perform exome target enrichment. The
captured library was sequenced on the Illumina HiSeq 2500 Sequencer with
paired-end 125 bp and mean coverage of 100X.
Variant
Calling and Filtering Raw data of
exome sequencing was filtered by removing adapter, contaminating reads and low
quality reads, and remains were the clean ones. The exome sequencing clean
reads were mapped to the reference human genome sequence (hg19)
(http://genome.ucsc.edu/) using the Burrows-Wheeler Alignment (BWA) tool, which
can do short reads alignment to a reference genome and support paired-end
mapping[26]. The sequence alignment/map (SAM)
file was then generated. Picard was used to mark and exclude the duplicate
reads. Variants [single nucleotide variants (SNVs), insertions and deletions]
calling was performed using the Genome Analysis Toolkit (GATK)[27] and MuTect software[28].
To
pinpoint the functionally important variants, the resulting SNVs were annotated
with ANNOVAR tool (http://www.openbioinformatics.org/annovar/)[29], and the information for variant frequencies and
location within genes were obtained. Moreover, the SNVs were sequentially
filtered and given higher priority with the following criteria: 1) minor allele
frequency (MAF) <0.01 in 1000 genomes project; 2) nonsynonymous SNVs; 3)
damaging as predicted by more than 4 bioinformatics programs (e.g. SIFT,
Polyphen2, LRT, MutationTaster, MutationAssessor, FATHMM, RadialSVM, LR); 4)
consistent with model of dominant disease transmission. Besides, more than 5X
coverage of the given positions were required for genotype calling.
Variant
Validation To validate
the variants identified through exome sequencing, candidate SNVs were selected
and sanger sequencing was performed at Majorbio (Shanghai, China). Peripheral
blood samples were obtained from additional 7 affected individuals and 3
unaffected individuals. Genomic DNA was extracted and SNVs were tested in the
original two individuals who underwent exome sequencing and ten additional
individuals. Oligonucleotide primers for PCR were designed by well-known
program Primer 3[30].
Silico
Analysis Protein
conservation was analyzed using the multiple alignment tools
(https://blast.ncbi.nlm.nih.gov/Blast.cgi). The affected residue was visualized
using MEGA7.0. The conserved domains present in the protein sequence were
identified using the Conserved Domain Search Service
(http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi).
RESULTS
Exome
Sequencing Identifies a Candidate Gene
The whole exomes of II:2 and III:1 were sequenced, followed by
variant detection and filtering. Totally, we generated 10.58 Gb and 20.18 Gb
raw sequences as paired-end 125 bp reads for II:2 and III:1, respectively.
After removing adapter, contaminating reads and low quality reads, 10.42 Gb and
19.85 Gb clean data were retained. Above 98% clean reads can be aligned to the
human reference sequence (Table 1). The exome sequencing led to the detection
of 525787 SNVs.
Table
1 Summary of exome sequencing data
|
Sample |
Raw data
(Gb) |
Clean data
(Gb) |
Map bases
rate (%) |
Target
region map bases (Gb) |
Target
region map bases rate (%) |
Coverage (%) |
Mean depth |
|
II:2 |
10.58 |
10.42 |
99.04 |
5.89 |
56.53 |
81.37 |
104.23 |
|
III:1 |
20.18 |
19.85 |
98.27 |
11 |
55.42 |
84.94 |
186.56 |
|
Average |
15.38 |
15.135 |
98.655 |
8.445 |
55.975 |
83.155 |
145.395 |
Considering
that a causal mutation is usually a rare variant or novel in the known
database, the SNVs with global MAF>0.01 in 1000 genomes project were excluded
and 111 738 SNVs were retained. Among the variants identified through exome
sequencing, we focused on the 1340 nonsynonymous SNVs in exonic region, which
can alter the coding sequence and were more likely associated with the disease.
With the assumption of dominant-inherited mode of the strabismus pedigree, 193
SNVs were retained which were shared by the two affected individuals. It is
well known that most pathogenic variants are predicted to be deleterious. Total
of eight bioinformatics programs were used to assess the likely functional
impact of nonsynonymous SNVs. Further filtering resulted in a list of 27 SNVs
that were damaging as predicted more than 4 bioinformatics programs (Table 2).
Given that strabismus is an eye development disease, we surveyed the literature
and narrowed down the gene list to two genes of PAX3 and MYO10 that may be
associated with strabismus.
Table 2 Deleterious rare variants (MAF<0.01) identified in the family
with strabismus
|
Chr |
Position |
Ref |
Var |
Gene |
Variant
type |
Amino acid
change |
1000
genome frequency |
EA-ESP frequency |
rs |
|
1 |
40422828 |
C |
T |
MFSD2A |
Nonsynonymous |
p.P55S |
0.000399 |
- |
rs181094032 |
|
1 |
45797401 |
G |
A |
MUTYH |
Nonsynonymous |
p.A345V |
0.001398 |
- |
rs35352891 |
|
2 |
74474313 |
C |
T |
SLC4A5 |
Nonsynonymous |
p.E637K |
- |
- |
- |
|
2 |
223160264 |
C |
A |
PAX3 |
Nonsynonymous |
p.R145L |
- |
- |
- |
|
3 |
156979081 |
G |
A |
VEPH1 |
Nonsynonymous |
p.R782C |
0.000399 |
- |
rs199678437 |
|
4 |
6302757 |
T |
C |
WFS1 |
Nonsynonymous |
p.V412A |
0.001398 |
- |
rs144951440 |
|
4 |
57340223 |
T |
C |
SRP72 |
Nonsynonymous |
p.Y120H |
0.0002 |
- |
- |
|
4 |
74442424 |
T |
A |
RASSF6 |
Nonsynonymous |
p.D215V |
0.000399 |
- |
rs200656717 |
|
4 |
103647776 |
C |
T |
MANBA |
Nonsynonymous |
p.S81N |
- |
- |
rs372866446 |
|
5 |
896841 |
C |
A |
TRIP13 |
Nonsynonymous |
p.P107H |
- |
- |
- |
|
5 |
16783553 |
C |
T |
MYO10 |
Nonsynonymous |
p.E165K |
- |
- |
- |
|
5 |
96329584 |
G |
T |
LNPEP |
Nonsynonymous |
p.R439L |
0.0002 |
- |
- |
|
6 |
75804894 |
C |
G |
COL12A1 |
Nonsynonymous |
p.G1696A |
- |
- |
- |
|
8 |
33449641 |
C |
T |
DUSP26 |
Nonsynonymous |
p.V176M |
0.0002 |
- |
- |
|
11 |
73717970 |
G |
A |
UCP3 |
Nonsynonymous |
p.R40C |
0.0002 |
0.000077 |
rs199727434 |
|
11 |
129795006 |
C |
T |
PRDM10 |
Nonsynonymous |
p.R464Q |
0.0002 |
0.0002 |
rs201242124 |
|
12 |
2224509 |
G |
A |
CACNA1C |
Nonsynonymous |
p.D57N |
- |
- |
- |
|
14 |
88946042 |
G |
A |
PTPN21 |
Nonsynonymous |
p.T578M |
- |
- |
- |
|
15 |
43132561 |
C |
A |
TTBK2 |
Nonsynonymous |
p.L96F |
- |
- |
- |
|
16 |
87885411 |
G |
A |
SLC7A5 |
Nonsynonymous |
p.R195W |
- |
- |
- |
|
17 |
3957414 |
G |
A |
ZZEF1 |
Nonsynonymous |
p.P1791S |
- |
- |
- |
|
17 |
63156387 |
G |
T |
RGS9 |
Nonsynonymous |
p.G81V |
- |
- |
- |
|
17 |
66890377 |
A |
T |
ABCA8 |
Nonsynonymous |
p.N991K |
0.0002 |
- |
- |
|
19 |
38103754 |
T |
C |
ZNF540 |
Nonsynonymous |
p.C525R |
0.000599 |
0.0005 |
rs138665562 |
|
19 |
50796922 |
G |
A |
MYH14 |
Nonsynonymous |
p.R1775H |
- |
0.000077 |
rs201923258 |
|
22 |
40801217 |
C |
T |
SGSM3 |
Nonsynonymous |
p.R120C |
- |
- |
- |
|
X |
43652695 |
T |
A |
MAOB |
Nonsynonymous |
p.Y300F |
- |
- |
- |
MAF:
Minor allele frequency; Chr: Chromosome; Ref: Reference allele; Var: Variant
allele; EA-ESP: European American Exome Sequencing Project; rs: Accession
number in dbSNP138.
MYO10
encodes a member of the myosin superfamily. Myosins are actin-dependent
molecular motors that play important roles in muscle contraction. The head
domain is a molecular motor, which utilizes ATP hydrolysis to generate directed
movement toward the plus end along actin filaments. A cyclical interaction
between myosin and actin provided the driving force for movement of the
extraocular muscles[31-32].
The mutation of c.493G>A in MYO10 (p.E165K) was highly conserved and the
altered amino acid residue (p.E165K) was located in the conserved motor domain.
Even so, the association of MYO10 and strabismus has not been reported.
Therefore, the candidate mutant in MYO10 was further excluded.
PAX3
is a member of the PAX family of transcription factors, which play critical
roles during fetal development. Mutations in PAX3 were associated with
Waardenburg syndrome with strabismus, and associated with
craniofacial-deafness-hand syndrome with short palpebral fissures and
hypertelorism. Considering that, we speculated that the mutant c.G434T
(p.R145L) in PAX3 was the most likely causative gene mutant in this Chinese
strabismus pedigree. The mutation of c.G434T in PAX3 was highly conserved
(Figure 1B) and the altered amino acid residue (p.R145L) was located in the
conserved PAX domain (Figure 1C).
Sanger
Sequencing of the Candidate Causative Variants To further
confirm the variant of c.434G>T in PAX3 in strabismus, Sanger sequencing was
performed in additional ten individuals. The results showed that the variants
were not observed in additional ten individuals with strabismus, strongly
supporting the genetic heterogeneity of strabismus.
DISCUSSION
Strabismus
was a large group of ophthalmic diseases with genetic heterogeneity among
families. Accumulating evidences have suggested that the etiology of strabismus
has important genetic factors[12-13,33]. While only the susceptibility locus 7p22.1 was
reported[16], leaving the genetic basis of this
disorder remains unclear and challenging. In the present study, we enrolled two
individuals with strabismus in a Chinese strabismus pedigree. In this pedigree,
the proposita and her mother were diagnosed as intermittent exotropia. We
suggested this was a congenital strabismus family and it was consistent with
the model of dominant disease transmission. Therefore, exome sequencing was
ideally suited to screen for the causal genes of the strabismus pedigree. Our
result identified a novel heterozygous mutation in PAX3 (c.G434T; p.R145L),
which was not reported in dbSNP 138, 1000 genome project or ESP6500. This
change may be associated with strabismus.
The
pathology of strabismus inheritance was complex[15,34-35]. In the current study, genetic
analysis was conducted on a Chinese strabismus pedigree, and a mutation in PAX3
was identified that may be responsible for hereditary susceptibility of
strabismus. PAX3 encoded a member of PAX family of transcription factors, which
played critical roles during fetal development. PAX3 gene contained 10 exons[36-37] and was mapped to chromosome
2q35[38]. The human PAX3 gene contained a PAX and
a paired-type homeobox. Molecular genetic studies were conducted and a series
of variations in the PAX3 gene were gradually identified in unrelated patients
or family patients with Waardenburg syndrome type 1[39-44]. In vitro functional expression studies showed
that the mutant proteins of PAX3 had decreased or abolished ability to
transactivate the MITF promoter[45]. Watanabe et
al[46] found that its paired domain or the
homeodomain failed to transactivate the MITF promoter, causing Waardenburg
syndrome in some individuals. Experiments on the mouse mutant splotch of
Waardenburg syndrome showed that mutations in PAX3 were associated with
Waardenburg syndrome that was related to human strabismus phenotypes[39,47-48]. Here, we
identified the conserved domains present in the protein sequence of PAX3 and
found that the candidate causal mutation of PAX3 (c.G434T; p.R145L) was located
in the conserved PAX domain. These findings supported our PAX3 variant as the
likely causative mutation, which may play roles in the pathological mechanism
of strabismus.
Ridgeway
and Skerjanc[49] suggested that strabismus was
associated with an imbalance between convergence and divergence. More evidences
indicated that ocular alignment depended on complex sensory, motor pathways,
and the development and function of the extraocular muscles. The expression of
PAX3 can control a cascade of transcriptional events, which are necessary for
myogenesis[49]. The absence of PAX3 can arrest
the muscle development[50]. PAX3/FKHR fusion
protein activated a myogenic transcription program involved in several aspects
of muscle function[51-53]. The
chimeric protein PAX3-FOXO1 was the most common genetic aberration in
rhabdomyosarcoma. Roeb et al[54] found
that myoblasts from transgenic mice expressing PAX3/FOXO1 were unable to
complete myogenic differentiation. A recent study reported that
oculo-auriculo-vertebral spectrum (OAVS) presented a generalized myopathy and
PAX3 may be responsible for non-branchiomeric myopathy with strabismus and limb
hypotrophy[55]. These findings suggested that
genetic aberrant of PAX3 may be involved in development and function of the
extraocular muscles, which affected the ocular alignment and contributed to
strabismus.
Taken
together, strabismus was a complex disease with significant genetic
heterogeneity, leading to the genetic findings hampered. We demonstrated the
presence of a novel causative mutation, c.434G-T (p.145R-L), in PAX3 in the
affected individuals, which may potentially contribute to strabismus
susceptibility. Further functional studies are needed to gain the pathogenic
mechanism and the role of PAX3 in strabismus.
There
was a limitation in this study. Herein, we only tested two subjects including
the proposita and her mother in the whole exomes sequencing. Other relatives
such as, father, uncle and the maternal grandparents of the proband should also
be studied. Additionally, larger numbers of sporadic individuals with
strabismus are needed to investigate the value of the identified variant.
Anyhow, we found the mutation gene of PAX3 in the strabismus family, which
provided a new field in understanding the genetic pathology of strabismus.
ACKNOWLEDGEMENTS
Conflicts
of Interest: Gong HM, None; Wang J, None; Xu J, None;
Zhou ZY, None; Li JW, None; Chen SF, None.
REFERENCES
2 Mezad-Koursh D, Leshno A, Ziv-Baran T, Stolovitch C. Refractive
changes induced by strabismus corrective surgery in adults. J Ophthalmol 2017;2017:2680204. [CrossRef] [PMC free article] [PubMed]
4 Rajamani M, Nagasubramanian V, Ayyavoo A, Raghupathy P, Dandapani R.
Surgically induced necrotizing scleritis following strabismus surgery treated
successfully with topical N-acetylcysteine in a child with congenital fibrosis
of extraocular muscles and Varadi Papp syndrome. Strabismus 2017:25(1):1-4. [CrossRef] [PubMed]
5 Engle EC. The genetic basis of complex strabismus. Pediatr Res 2006;59(3):343-348. [CrossRef] [PubMed]
6 Multi-ethnic Pediatric Eye Disease Study Group. Prevalence of
amblyopia and strabismus in African American and Hispanic children ages 6 to 72
months the multi-ethnic pediatric eye disease study. Ophthalmology 2008;115(7):1229-1236.e1. [CrossRef] [PMC free article] [PubMed]
7 Chia A, Dirani M, Chan YH, Gazzard G, Au Eong KG, Selvaraj P, Ling Y,
Quah BL, Young TL, Mitchell P, Varma R, Wong TY, Saw SM. Prevalence of
amblyopia and strabismus in young singaporean Chinese children. Invest Ophthalmol Vis Sci 2010;
51(3):3411-3417. [CrossRef] [PMC free article] [PubMed]
8 Matsuo T, Matsuo C. The prevalence of strabismus and amblyopia in
Japanese elementary school children. Ophthalmic
Epidemiol 2005;12(1): 31-36. [CrossRef] [PubMed]
10 Preising MN, Steinmuller PH, Lorenz B. Recruitment of suitable
families to identify causative genes in hereditary strabismus. Klin Monbl Augenheilkd 2015;232(10):1158-1164.
[PubMed]
11 McKean-Cowdin R, Cotter SA, Tarczy-Hornoch K, Wen G, Kim J, Borchert
M, Varma R. Prevalence of amblyopia or strabismus in Asian and non-Hispanic
White preschool children: multi-ethnic pediatric eye disease study. Ophthalmology 2013;120:2117-2124. [CrossRef] [PMC free article] [PubMed]
12 Paul TO, Hardage LK. The heritability of strabismus. Ophthalmic Genet 1994;15(1):1-18. [CrossRef] [PubMed]
13 Mash AJ, Spivey BE. Genetic aspects of strabismus. Doc Ophthalmol 1973;34(1):285-291. [CrossRef] [PubMed]
14 Maconachie GD, Gottlob I, McLean RJ. Risk factors and genetics in
common comitant strabismus: a systematic review of the literature. JAMA Ophthalmol 2013;131(9):1179-1186. [CrossRef] [PubMed]
15 Schlossman A, Priestley BS. Role of heredity in etiology and
treatment of strabismus. AMA Arch
Ophthalmol 1952;47:1-20. [CrossRef] [PubMed]
16 Parikh V, Shugart YY, Doheny KF, et
al. A strabismus susceptibility locus on chromosome 7p. Proc Natl Acad Sci U S A 2003;100(21):12283-12288.
[CrossRef] [PMC free article] [PubMed]
17 Rice A, Nsengimana J, Simmons IG, Toomes C, Hoole J, Willoughby CE, Cassidy
F, Williams GA, George ND, Sheridan E, Young TL, Hunter TI, Barrett BT, Elliott
DB, Bishop DT, Inglehearn CF. Replication of the recessive STBMS1 locus but
with dominant inheritance. Invest
Ophthalmol Vis Sci 2009;50(7):3210-3217. [CrossRef] [PubMed]
18 Khan AO, Shinwari J, Abu Dhaim N, Khalil D, Al Sharif L, Al Tassan N.
Potential linkage of different phenotypic forms of childhood strabismus to a
recessive susceptibility locus (16p13.12-p12.3). Mol Vis 2011;17:971-976. [PMC free article] [PubMed]
19 Shaaban S, Matsuo T, Fujiwara H, Itoshima E, Furuse T, Hasebe S,
Zhang Q, Ott J, Ohtsuki H. Chromosomes 4q28.3 and 7q31.2 as new susceptibility
loci for comitant strabismus. Invest
Ophthalmol Vis Sci 2009;50(2):654-661. [CrossRef] [PubMed]
20 Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer
T, Wong M, Bhattacharjee A, Eichler EE, Bamshad M, Nickerson DA, Shendure J.
Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009;461:272-276. [CrossRef] [PMC free article] [PubMed]
21 Wang JL, Yang X, Xia K, Hu ZM, Weng L, Jin X, Jiang H, Zhang P, Shen
L, Guo JF, Li N, Li YR, Lei LF, Zhou J, Du J, Zhou YF, Pan Q, Wang J, Wang J,
Li RQ, Tang BS. TGM6 identified as a novel causative gene of spinocerebellar
ataxias using exome sequencing. Brain 2010;133(Pt
12):3510-3518. [CrossRef] [PubMed]
22 Alazami AM, Hijazi H, Al-Dosari MS, Shaheen R, Hashem A, Aldahmesh
MA, Mohamed JY, Kentab A, Salih MA, Awaji A, Masoodi TA, Alkuraya FS. Mutation
in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes
intellectual disability and strabismus. J
Med Genet 2013;50:425-430. [CrossRef] [PubMed]
23 Baschal EE, Wethey CI, Swindle K, Baschal RM, Gowan K, Tang NL,
Alvarado DM, Haller GE, Dobbs MB, Taylor MR, Gurnett CA, Jones KL, Miller NH.
Exome sequencing identifies a rare HSPG2 variant associated with familial
idiopathic scoliosis. G3 (Bethesda) 2014;5(2):167-174.
[CrossRef] [PMC free article] [PubMed]
24 Cao Y, Gao Z, Li L, Jiang X, Shan A, Cai J, Peng Y, Li Y, Jiang X,
Huang X, Wang J, Wei Q, Qin G, Zhao J, Jin X, Liu L, Li Y, Wang W, Wang J, Ning
G. Whole exome sequencing of insulinoma reveals recurrent T372R mutations in
YY1. Nat Commun 2013;4:2810. [CrossRef] [PubMed]
25 Qiao D, Lange C, Laird NM, Won S, Hersh CP, Morrow J, Hobbs BD, Lutz
SM, Ruczinski I, Beaty TH, Silverman EK, Cho MH. Gene-based segregation method
for identifying rare variants in family-based sequencing studies. Genet Epidemiol 2017;41(4):309-319. [CrossRef] [PubMed]
26 Li H, Durbin R. Fast and accurate short read alignment with
Burrows-Wheeler transform. Bioinformatics
2009;25(14):1754-1760. [CrossRef] [PMC free article] [PubMed]
27 McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A,
Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis
Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing
data. Genome Res 2010;20(9):1297-1303.
[CrossRef] [PMC free article] [PubMed]
28 Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez
C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic
point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31(3):213-219. [CrossRef] [PMC free article] [PubMed]
29 Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic
variants from high-throughput sequencing data. Nucleic Acids Res 2010;38(16):e164. [CrossRef] [PMC free article] [PubMed]
30 Krawetz SA, Womble DD. Design and implementation of an introductory
course for computer applications in molecular genetics. A case study. Mol Biotechnol 2001;17(1):27-41. [CrossRef]
31 Pelouch V. Molecular aspects of regulation of cardiac contraction. Physiol Res 1995;44(1):53-60. [PubMed]
32 Yao J, Wang X, Ren H, Liu G, Lu P. Ultrastructure of medial rectus
muscles in patients with intermittent exotropia. Eye (Lond) 2016;30(1):146-151. [CrossRef] [PMC free article] [PubMed]
33 Cantolino SJ, Von Noorden GK. Heredity in microtropia. Arch Ophthalmol 1969;81(6):753-757. [CrossRef] [PubMed]
34 Hegmann JP, Mash AJ, Spivey BE. Genetic analysis of human visual
parameters in populations with varying incidences of strabismus. Am J Hum Genet 1974;26(5):549-562. [PMC free article] [PubMed]
35 Maumenee IH, Alston A, Mets MB, Flynn JT, Mitchell TN, Beaty TH.
Inheritance of congenital esotropia. Trans
Am Ophthalmol Soc 1986;84: 85-93. [PMC free article] [PubMed]
36 Barber TD, Barber MC, Cloutier TE, Friedman TB. PAX3 gene structure,
alternative splicing and evolution. Gene 1999;237(2):311-319.
[CrossRef]
37 Jalilian N, Tabatabaiefar MA, Farhadi M, Bahrami T, Noori-Daloii MR.
A novel mutation in the PAX3 gene causes Waardenburg syndrome type I in an
Iranian family. Int J Pediatr
Otorhinolaryngol 2015;79(10): 1736-1740. [CrossRef] [PubMed]
38 Ishikiriyama S. Gene for Waardenburg syndrome type I is located at
2q35, not at 2q37.3. Am J Med Genet 1993;46(5):608.
[CrossRef] [PubMed]
39 Tassabehji M, Read AP, Newton VE, Harris R, Balling R, Gruss P,
Strachan T. Waardenburg's syndrome patients have mutations in the human
homologue of the Pax-3 paired box gene. Nature
1992;355(6361):635-636. [CrossRef] [PubMed]
40 Baldwin CT, Hoth CF, Macina RA, Milunsky A. Mutations in PAX3 that
cause Waardenburg syndrome type I: ten new mutations and review of the
literature. Am J Med Genet 1995;58(2):115-122.
[CrossRef] [PubMed]
41 Farrer LA, Arnos KS, Asher JH Jr, Baldwin CT, Diehl SR, Friedman TB,
Greenberg J, Grundfast KM, Hoth C, Lalwani AK, et al. Locus heterogeneity for
Waardenburg syndrome is predictive of clinical subtypes. Am J Hum Genet 1994;55(4):728-737. [PMC free article] [PubMed]
42 Tassabehji M, Newton VE, Liu XZ, Brady A, Donnai D, Krajewska-Walasek
M, Murday V, Norman A, Obersztyn E, Reardon W, et al. The mutational spectrum
in Waardenburg syndrome. Hum Mol Genet 1995;4(11):2131-2137.
[CrossRef]
43 Zlotogora J, Lerer I, Bar-David S, Ergaz Z, Abeliovich D.
Homozygosity for Waardenburg syndrome. Am
J Hum Genet 1995;56(5):1173-1178. [PMC free article] [PubMed]
44 Chen H, Jiang L, Xie Z, Mei L, He C, Hu Z, Xia K, Feng Y. Novel
mutations of PAX3, MITF, and SOX10 genes in Chinese patients with type I or
type II Waardenburg syndrome. Biochem
Biophys Res Commun 2010;397(1):70-74. [CrossRef] [PubMed]
45 Zhang H, Chen H, Luo H, An J, Sun L, Mei L, He C, Jiang L, Jiang W,
Xia K, Li JD, Feng Y. Functional analysis of Waardenburg syndrome-associated
PAX3 and SOX10 mutations: report of a dominant-negative SOX10 mutation in
Waardenburg syndrome type II. Hum Genet 2012;
131(3):491-503. [CrossRef] [PubMed]
46 Watanabe A, Takeda K, Ploplis B, Tachibana M. Epistatic relationship
between Waardenburg syndrome genes MITF and PAX3. Nat Genet 1998;18(3):283-286. [CrossRef] [PubMed]
47 Tassabehji M, Newton VE, Leverton K, Turnbull K, Seemanova E, Kunze
J, Sperling K, Strachan T, Read AP. PAX3 gene structure and mutations: close
analogies between Waardenburg syndrome and the Splotch mouse. Hum Mol Genet 1994;3(7):1069-1074. [CrossRef]
48 Morell R, Friedman TB, Moeljopawiro S, Hartono, Soewito, Asher JH Jr.
A frameshift mutation in the HuP2 paired domain of the probable human homolog
of murine Pax-3 is responsible for Waardenburg syndrome type 1 in an Indonesian
family. Hum Mol Genet 1992;1(4):243-247.
[CrossRef]
49 Ridgeway AG, Skerjanc IS. Pax3 is essential for skeletal myogenesis
and the expression of Six1 and Eya2. J
Biol Chem 2001;276(22): 19033-19039. [CrossRef] [PubMed]
50 Relaix F, Rocancourt D, Mansouri A, Buckingham M. A
Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 2005;435(7044):948-953. [CrossRef] [PubMed]
51 Sublett JE, Jeon IS, Shapiro DN. The alveolar rhabdomyosarcoma
PAX3/FKHR fusion protein is a transcriptional activator. Oncogene 1995;11(3):545-552. [PubMed]
52 Scheidler S, Fredericks WJ, Rauscher FJ 3rd, Barr FG, Vogt PK. The
hybrid PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma transforms fibroblasts
in culture. Proc Natl Acad Sci U S A 1996;93(18):
9805-9809. [CrossRef]
53 Khan J, Bittner ML, Saal LH, Teichmann U, Azorsa DO, Gooden GC, Pavan
WJ, Trent JM, Meltzer PS. cDNA microarrays detect activation of a myogenic
transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci U S A 1999;96(23):13264-13269. [CrossRef]
54 Roeb W, Boyer A, Cavenee WK, Arden KC. PAX3-FOXO1 controls expression
of the p57Kip2 cell-cycle regulator through degradation of EGR1. Proc Natl Acad Sci U S A 2007;104(46):18085-18090.
[CrossRef] [PMC free article] [PubMed]
55 Murialdo G, Piazzi A, Badolati G, Calcagno E, Berio A.
Oculo-auriculo-vertebral spectrum with myopathy and velopharyngeal
insufficiency. A case report with a non-branchiomeric muscle biopsy. Pediatr Med Chir 2016;38(2):121. [CrossRef] [PubMed]