
・Review・ Current
Issue IF in JCR CiteScore ・Submission・ In Press Recent Accepted PMC RSS
Citation: Singh M, Tyagi SC. Metalloproteinases as mediators of inflammation and
the eyes: molecular genetic underpinnings governing ocular pathophysiology. Int
J Ophthalmol
2017;10(8):1308-1318
Metalloproteinases
as mediators of inflammation and the eyes:
molecular genetic underpinnings governing ocular pathophysiology
Mahavir Singh, Suresh C Tyagi
Eye
and Vision Science Laboratory, Department of Physiology, University of
Louisville School of Medicine, Louisville, KY 40202, USA
Correspondence
to: Mahavir Singh. Eye and Vision Science Laboratory, Department of
Physiology, University of Louisville School of Medicine, Louisville, KY 40202,
USA. mahavir.singh@louisville.edu; gene2genetics@gmail.com
Received:
2016-12-22
Accepted: 2017-06-01
Abstract
There
are many vision threatening diseases of the eye affecting millions of people
worldwide. In this article, we are summarizing potential role of various matrix
metalloproteinases (MMPs); the Zn (2+)-dependent endoproteases in eye health
along with pathogenesis of prominent ocular diseases such as macular
degeneration, diabetic retinopathy, and glaucoma via understanding MMPs
regulation in affected patients, interactions of MMPs with their substrate
molecules, and key regulatory functions of tissue inhibitor of
metalloproteinases (TIMPs) towards maintaining overall homeostasis.
KEYWORDS: age-related
macular degeneration; choroidal neovascularization; diabetes; glaucoma;
metalloproteinases; tissue inhibitors of metalloproteinases
DOI:10.18240/ijo.2017.08.20
Citation: Singh M, Tyagi SC. Metalloproteinases as mediators of inflammation and
the eyes: molecular genetic underpinnings governing ocular pathophysiology. Int
J Ophthalmol
2017;10(8):1308-1318
INTRODUCTION
Matrix
metalloproteinases (MMPs) constitute a large family of secreted and membrane
associated zinc-dependent proteolytic endopeptidases. They are known to perform
roles in regulation of tissue morphogenesis, motility, cell growth, response to
injury, and extracellular matrix (ECM) remodeling not only by degrading matrix
related proteins, but also through well-controlled proteolysis of specific
extracellular targets that includes receptors, cytokines, growth factors, and
adhesion molecules[1-2].
Proteolytic activities of MMPs are regulated at various levels and they are
produced mainly as zymogens requiring activation by dedicated proteases[3-4]. And once activated, MMPs’
activities are often controlled by their endogenous inhibitors; known as tissue
inhibitors of metalloproteinases (TIMPs) (Figure 1)[5-6]. Literature survey reveals that MMPs when dysregulated
can participate in a variety of medical conditions since they serve as crucial
players in cell proliferation, differentiation, angiogenesis, apoptosis, and
immune defense[7-8]. Thus,
functional dysregulation as accompanied by excessive activation of MMPs have
been associated with many human diseases[1,7,9-11]. For example, MMP-10 can degrade
a broad spectrum of matrix proteins[12], and
activate MMP-1, 7, 8, and 9[13]. MMPs are present
in almost all tissues impacting many aspects of their unique physiology[14-15]. Like in other organs MMPs are
also responsible for maintenance and remodeling of ocular architecture by
influencing a wide range of processes in eyes. Their substrates represent a
variety of ECM components such as cytokines, chemokines and cell surface
molecules. MMPs and TIMPs have been shown to co-localize in human
inter-photoreceptor matrix (IPM)[15] playing an
important role in physiological reconstruction and turnover of IPM. They are
also responsible during embryogenesis, angiogenesis, and ocular wound healing[16]. However, they can be detrimental in breaking apart
basement membrane contributing to ulcerations such as those observed in corneal
tissue[17].
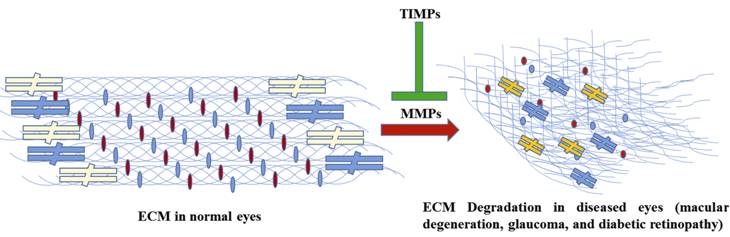
Figure 1 A simple schematic highlighting the effects of MMPs on ocular
ECM metabolism During disease conditions the homeostatic balance between MMPs and TIMPs
gets disturbed leading to degradation of ECM normal architecture in the eyes of
the affected patients.
Over the last 30y our lab has been actively studying mechanisms that
govern maintenance of tissues’ structural integrity[18],
post-transcriptional regulation and metabolism of MMPs and TIMPs in human heart
failure, atherosclerosis, microvascular permeability and vascular diseases[19-24], involvement of MMPs in
diabetes, hypertension, oxidative stress, nephropathy, and autophagy[25-28]. From our experience, we know
that endogenous TIMPs regulate proteolytic activity by binding tightly to MMPs’
active sites. While TIMPs can inhibit most if not all MMPs, available data also
reveal tremendous heterogeneity in their affinities of different TIMP/MMP
pairs, and that the structural features which differentiate stronger from
weaker complexes are not well understood. Like MMPs, TIMPs represent
multifunctional proteins having activities that are mediated through protein-protein
interactions (PPIs) with other partners. For example, TIMP-2 has been reported
to inhibit almost all MMPs that have been studied[5-6] and as listed in the MEROPS; a peptide database[29] including MMP-1, 2, 3, 7, 8, 9, 10, 13, 19, MT1-MMP,
MT2-MMP, MT3-MMP, MT4-MMP, and MT6-MMP, as well as ADAM12[30].
As far as physical nature of their partnerships goes the respective inhibition
constants (Ki) for their
interactions vary widely from 0.6 fmol/L for full-length MMP-2[31] to 5.8 nmol/L for MMP-10’s catalytic domain[32]. Also, TIMP-2 can interact with α3β1 integrin molecule and regulate cell cycle progression and angiogenesis
process via MMP-independent mechanisms[5-6,33]. The general structural basis for
inhibition of MMPs by TIMPs was successfully revealed in crystal structures of
MMP-3/TIMP-1[34] and MT1-MMP/TIMP-2 complexes[35], and subsequently expanded with later structures of
MMP-13/TIMP-2[36] and MMP-10/TIMP-1 complexes[32], along with complexes of MMP-1 and MT1-MMP with
N-terminal domain of TIMP-1, which makes majority of intermolecular contacts[37-38]. At a given time in healthy
tissue, MMPs and TIMPs levels are found in the stoichiometric ratio of 1:1.
Whenever there is an excess MMP it can lead to tissue degradation and the same
is true about excess amount of TIMP which may result into ECM accumulation.
Thus, the balance needs to be maintained for the proper functioning of the
tissue otherwise it may result into an altered regulation of ECM remodeling
(such as scarring). In short, MMPs’ activities are carefully controlled by
TIMPs, and ultimately balance between MMPs and TIMPs determines the final
outcomes in a particular tissue (Figure 1). In glaucoma patients, there seems
to be an imbalance between MMPs and TIMPs in the eye’s chamber angle playing a
role in the pathogenesis of the disease itself. Similarly, imbalance in TIMPs’
favor can promote initiation of fibrosis leading to tissue remodeling as seen
in case of MMP-9 which was shown to be important in corneal stromal remodeling
in humans[39] and at the same time its
involvement in corneal injury as reported in a study which was conducted on
rats[40]. This study attempts to review an
ever-expanding literature on molecular genetics aspects of MMPs and their
related biology along with a select description in important ocular diseases
such as macular degeneration, diabetic retinopathy (DR) and glaucoma that
affect millions of people around the world.
Information About Diseases in Detail
Matrix metalloproteinases in macular
degeneration Age-related macular degeneration (AMD)
leads to adverse vascular changes and is the most common cause of irreversible
vision loss in elderly people globally. It may result from degeneration of rods
and cones in the macular region of central retina which is responsible for high
acuity vision. Death of photoreceptors appears to be a direct consequence of
degeneration of neighboring retinal pigment epithelium (RPE) cells. Drusen
formation; abnormal deposits in ECM, is an important hallmark of AMD disease.
Typically, drusen lie between RPE basement membrane and inner collagenous layer
of Bruch's membrane (BM) and contain ECM along with other molecules. It is
hypothesized that drusen may result from the failure to dispose off RPE-derived
molecules such as ECM, or it may be the result of dysregulated inflammatory
immune mediators. Proinflammatory cytokines were recently reported to decrease
the expression of genes that are critical for normal functioning of RPE[41-42]. MMP-9 has been shown to
participate in the development of choroidal neovascularization (CNV) as part of
AMD pathogenesis[43-45].
Although the etiology of AMD is multifactorial[44,46-47] but a significant role is
played by MMP-1, 2, 9, 14 and TIMP-3. It became evident that a continuous
rebuilding of ECM occurs in the early and advanced AMD disease simultaneously
with the combined malfunctioning of RPE and endothelial cells. Pathological
degradation or accumulation of ECM structural components are usually caused by
impairment or hyperactivity of specific MMPs/TIMPs interactions, and is also
influenced by genetic and environmental factors. Fiotti et al[48] observed a relationship between polymorphisms in
MMP-9 and CNV. More recently, it was shown that MMP-9 rs3918242 (C>T) single
nucleotide polymorphism (SNP) was found to play a role in AMD development, and
the effect was more pronounced in patients who were less than 65 years of age
(Table 1)[49-53].
Table 1 Genes or variants associated with eye diseases
|
No. |
Disease |
Gene names |
References |
|
1 |
AMD/Sorby’s
fundus dystrophy |
SNPs/mutations
in MMP-2, 3, 9, TIMP-2, 3, ER-α |
MMP-9: Fiotti et al (2005); TIMP-3: Langton et al (2000); MMP-2, 3, 9: Liutkeviciene et al (2015); MMP-2, TIMP-2: Ortak et al (2013); ER-α, MMP-2: Seitzman et al (2008); TIMP-3: Weber et al (1994); TIMP-3: Chen et al (2010) |
|
2 |
DR/optic
disc anomaly |
SNPs in
MMP-2, 9, VEGF, SDH and a 6-Kbp heterozygous triplication upstream of MMP-19
regulatory sequences |
MMP-2,
9: Beranek et al (2008); MMP-9:
Maeda et al (2001); MMP-18:
Hazlewood et al (2015); VEGF,
SDH: Rusin and Majsterek (2007); MMP-2:
Yang et al (2010) |
|
3 |
Glaucoma |
SNPs in
MMP-1, 2, 3, 9, 12, 16, IL-1beta, TIMP-1, PTGFR |
MMP-1,
9, 12, IL-1beta and TIMP-1: Markiewicz
et al (2013); MMP-9,
PTGFR: Zhang et al (2016); MMP-1,
2, 3, 16: Vranka et al (2015) |
Only select information is listed. AMD: Age-related macular
degeneration; DR: Diabetic retinopathy; SNP: Single nucleotide polymorphism;
MMP: Matrix metalloproteinases; TIMP: Tissue inhibitors of metalloproteinases;
ER: Estrogen receptor; VEGF: Vesicular endothelial growth factor; SDH: Sorbitol
dehydrogenase; PTGFR: Prostaglandin F2α receptor
gene.
Interestingly, circulating MMPs and TIMPs have been suggested to
participate in a variety of vascular remodeling and angiogenesis processes[54] but it is still not clear whether these circulating
MMPs are linked to AMD pathogenesis or not.
Mutations in TIMP-3 cause Sorsby fundus dystrophy, another blinding
disease with similarities to AMD[55]. TIMP-3 is a
component of BM[56-57] and is
found to be concentrated in drusen[58] which are
cold spots for proteolysis activity. Chau et al[59]
reported a connection between elevated plasma MMP-9 levels and AMD. By
contrast, Zeng et al[60] reported a link
between increased levels of circulating gelatinases (MMP-2 and MMP-9) and
polypoidal choroidal vasculopathy (PCV)[61]; an
abnormal choroidal vasculopathy distinct from typical CNV[62]
but not AMD. However, in AMD lesions, it is believed that activated endothelial
cells release MMPs and destroy the BM[63]. It has
been reported that total levels of active MMP-2 and MMP-9 were significantly
reduced in BM-choroid preparations from AMD patients[64].
Paradoxically, downregulation of MMP-2 and MMP-9 may lead to advanced
pathological changes that can progress to AMD but Ahir et al[65] demonstrated that administration of activated MMP-2
and MMP-9 can improve fluid permeability of BM. As per their findings it
appears that reactivation of MMP pathway may be an effective therapeutic modality
in AMD patients. Along these lines, it is worth to mention that “nano-second”
laser which uses pulse durations in the range of nanosecond restricts heat
transients to <30 μmol/L can specifically target RPE cells
without any damage to photoreceptors or BM[66].
Similarly, Zhang et al[67] corroborated
nano-second laser induced RPE-mediated release of MMP enzymes. Subsequent
findings from one-year clinical trial using ultrashort-pulse laser has been
reported and it is interesting to note that a single application of laser to
macula of AMD patients can improve the overall macular appearance and its
functions. These observations reflected the results that unilateral laser
photocoagulation can induce a bilateral MMP pathway activation by releasing a
host of circulating factors. Taken together, targeting MMP activation may be a
useful approach for the treatment of AMD disease which could serve as a
potential therapeutic option for patients.
Matrix metalloproteinases in diabetic
eyes DR is one of the most dreaded complications in diabetic patients which
can lead to both severe forms of vasculopathy and neuropathy. Despite extensive
progress in diabetic research, the mechanism(s) responsible for development of
this chronic disease remains unknown. It is characterized by endothelial
malfunctioning, enhanced permeability of vascular structures, hemostatic
abnormalities, tissue ischemia, and neo-angiogenesis[68]. Several genes (MMP-2, 9, 19) and
their variants have been linked in pathogenesis of DR along with epigenetic and
environmental factors (Table 1)[69-73]. Several pathways are known to be
involved in regulation of epigenetic changes such as micro-ribonucleic acids
(microRNAs), DNA methylation, and histone acetylation[74]. In diabetic patients MMPs degrade ECM and thus dysregulate many cellular
functions via signaling stress responses in the eye. Retina and macula
are highly prone to free radical mediated damage, and this can have a
devastating effect on one’s vision. Oxidative stress due to excessive
production of reactive oxygen species (ROS) can overwhelm intrinsic antioxidant
capacity of cells and thus can induce injury to tissues[75]
including cells in ocular and other compartments. Thus, oxidative stress
affects the development of DR. MMP-2, a most ubiquitous member of MMP family has
been shown to be a potent sensitizer for oxidative stress. When effects of
mitochondrial superoxide scavenger on glucose-induced alterations in MMP-2, and
its proenzyme activator MT1-MMP and its physiological inhibitor TIMP-2 were
determined in retinal endothelial cells, it was revealed that glucose induced
activation of retinal capillary cell MMP-2 and MT1-MMP and a concomitant
decrease in TIMP-2 suggesting a possible use of MMP-2 targeted therapy to
inhibit the development of DR[76].
Diabetes alters the blood-retinal barrier (BRB) possibly by via breakdown
of endothelial cell tight junctions. To prove this Giebel et al[77] studied expression of extracellular proteinases in an
animal model of early DR and MMPs’ expression was
investigated in retinas of rats with 12wk of diabetes. In this study role of MMPs in regulating tight junction function was
investigated in retinal endothelial and RPE cells by measuring transepithelial
electrical resistance (TER). Retinas of diabetic
animals demonstrated elevated levels of MMP-2, MMP-9 and MMP-14 messenger RNAs.
A significant increase in production of MMP-9 was seen when cells were exposed
to high glucose conditions. Both cell types treated with purified MMP-2 or MMP-9
were found to have alterations of tight junction functions as shown by
decreased TER. Western blot analysis of cell extracts treated with MMPs (MMP-2
or MMP-9), revealed specific degradation of tight junction protein, occludin.
Results suggested that elevated expression of MMPs
in retina may facilitate an increase in vascular permeability by a mechanism
involving proteolytic degradation of tight junction protein occludin followed
by disruption of overall tight junction complex. Similarly, when levels of both MMP-2 and MMP-9 in vitreous samples
collected from proliferative diabetic retinopathy
(PDR) patients were examined, ProMMP-9 and activated MMP-9 amounts were
significantly increased in patients. In addition, TIMP-1 levels were also
increased in PDR patients and functionally inhibited activation of MMP-9 in
vitreous samples. These results clearly indicated that activated MMP-9 might be
involved in hemorrhagic transformation in patients affected with PDR[78].
Recent findings demonstrate that pathogenesis of DR involves
H-Ras and MMP-9 acting in concert to accelerate apoptosis of cells such as
retinal capillary cells. Using isolated retinal endothelial cells, effect of
regulation of H-Ras downstream signaling cascade, Raf-1, MEK, and ERK, was
investigated on glucose-induced activation of MMP-9 and the results confirmed
that DR
increased MMP-9 activity in retinal micro-vessels; the site associated with DR, and it was
also accompanied by activated H-Ras signaling pathway (Raf-1/ERK). Together,
these findings suggest that Ras/Raf-1/MEK/ERK cascade plays an important role
in the activation of retinal MMP-9 resulting in apoptosis of capillary cells.
Therefore, understanding the upstream mechanisms which are responsible for
activation of MMP-9 should be helpful in identifying novel molecular targets
for future pharmacological interventions to inhibit development and progression
of DR in
vulnerable patient populations[79-80].
In another study, it was reported that active PDR patients express MMP-9 again
suggesting that MMP-9 is one of the important factors in progression of PDR[81]. When fibrovascular membranes from PDR subjects were
analyzed they stained positive for MMP-1, MMP-2, MMP-3, MMP-9, TIMP-1, TIMP-2,
and TIMP-3 and there was a characteristic staining for MMP-9 within the
perivascular matrix of PDR membranes. These and
related findings indicate that MMPs are involved
in degradation of fibrovascular tissue matrix, as
well as TIMP-1 and TIMP-2, are found in a large proportion of membranes
suggesting the existence of common pathways of ECM degradation in pathological
processes leading to retinal neovascularization and fibrosis[82].
Using human donor corneal tissues researchers
wanted to know whether changes in corneal structural components might be caused
by decreased gene activities or increased degradation process. Expression
levels of α-1, α-5, and β-1 laminin chains; nidogen-1/entactin;
integrin α-3 and β-1 chains in diabetic and DR corneal
epithelium were like normal meaning that the observed basement membrane and
integrin alterations were unlikely to occur because of a decreased synthesis.
mRNA quantities of matrix MMP-10/stromelysin-2
were significantly increased in DR corneal epithelium and stroma, and of
MMP-3/stromelysin-1 in DR corneal stroma. mRNA levels of five other proteinases
and of three tissue inhibitors of MMPs were like
normal in diabetic and DR corneal epithelium and
stroma. The resultant data suggested that changes in laminins, nidogen-1/entactin,
and epithelial integrin in DR corneas may occur because of an increased
proteolytic degradation.
Overexpression of MMP-10 in diabetic
corneal epithelium seemed to be the major contributor towards observed changes
in DR corneas. Such changes may bring epithelial adhesive abnormalities as seen
in diabetic corneas[83].
While studying effects of posterior vitreous detachment (PVD), proliferative
membrane, vitreous hemorrhage, traction detachment, and cystoid macular edema
on MMP activities in human vitreous samples from
patients with DR and other vitreoretinal diseases Jin et al[84] discovered that MMP-9 may be involved in DR and that
partial PVD may be related to MMP-9 activity in DR. Out of many MMPs examined
in vitreous samples, only levels of MMP-2 and MMP-9 were significantly higher
in PDR than control subjects. Immunohistochemical study also demonstrated
localization of MMP-2 and MMP-9 in endothelial cells and glial cells of
fibrovascular tissues. Here, MMP-2 was colocalized with MT1-MMP and TIMP-2,
which are an activator and an activation-enhancing factor respectively for
proMMP-2 clearly demonstrating that proMMP-2 is efficiently activated in the
fibrovascular tissues of PDR patients, probably via interaction with
MT1-MMP and TIMP-2. These observations suggest the possibility that activities
of MMP-2 and MT1-MMP are involved in the formation of fibrovascular tissues
alterations[85].
Matrix metalloproteinases in
glaucoma Elevated intraocular pressure (IOP) is a primary risk factor for the
etiology of glaucoma and primary open angle glaucoma (POAG) is the main cause
of irreversible blindness in people all over the world. MMPs and their
regulators; TIMPs and interleukins (ILs) have been extensively studied as POAG
risk factors. Lowering IOP remains the only effective treatment for patients
with glaucoma. Trabecular meshwork (TM) in concert with ciliary muscle
contraction and relaxation provide a working control of aqueous humor outflow
in our eyes. In doing so, TM plays an important physiological role in
regulating IOP which is predominantly mediated by cytoskeletal and
contractility mechanisms as well as signal transduction pathways. This complex
system is subject to alteration as one ages and during progression of glaucoma
disease. Factors such as a compromised antioxidant defense system[75] and altered ECM metabolism are known to contribute to
impaired aqueous humor outflow which is common in POAG, exfoliation syndrome,
and exfoliation glaucoma (XFG).
While IOP is a well-known predisposing risk factor as mentioned earlier
for glaucoma, the etiology of glaucomatous optic neuropathy (GON) is currently
not well understood. It appears that a variety of other potential factors
particularly those of a vascular nature might be at play in GON because an
unstable oxygen content supply as opposed to prevalence of chronic hypoxic
conditions seems to contribute to the etiology of GON resulting from constant
fluctuations in local oxygen tension leading to an unstable ocular blood flow
(OBF) which in turn can fluctuate if IOP spikes or blood pressure drops. In
such a scenario OBF autoregulation becomes defective because the main reason
for disturbed autoregulation is a primary vascular dysregulation (PVD),
particularly in the context of the so-called Flammer syndrome. This unstable
oxygen tension can therefore lead to local oxidative stress with many
detrimental effects such as activation of glial cells, which can alter their
morphology and gene expression pattern. Because of these changes, the local
concentrations of nitric oxide (NO2) and MMPs increase leading to
remodeling via digestion of ECM components. Short-lived NO2 can
easily diffuse into neighboring axons, allowing a fusion with superoxide anion
and thus generates a cell damaging peroxynitrite. These developments can
further contribute to the development and progression of GON phenotype[86].
Since TM in anterior chamber of the eye regulates IOP by generating
resistance to aqueous humor outflow so IOP can result from reduced aqueous
humor outflow. TM consists of specialized cells within a complex ECM
environment. An imbalance between ECM-degrading MMPs and TIMPs within TM is
thought to contribute to POAG (Figure 1). Quantification of TIMPs and MMPs
levels in aqueous humor from glaucomatous and non-glaucomatous patients did
reveal an imbalance among MMPs and TIMPs with a shift toward raised TIMP
levels. This shift may result in the inhibition of MMPs activities, leading to
an altered ECM composition in TM and thereby contributing to increased outflow resistance[87]. Researchers have also observed that aqueous humor
obtained from patients with POAG display increased expression of MMP-1, MMP-9,
and MMP-12 in comparison to control samples. Recent studies showed involvement
of several SNPs for MMPs, TIMPs and ILs encoding genes in patients with POAG.
Investigative association of -1607 1G/2G MMP-1, -1562 C/T MMP-9, -82 A/G
MMP-12, -511 C/T IL-1β and 372 T/C TIMP-1 confirmed
statistically significant increase in POAG development risk of -1607 2G/2G
MMP-1 genotype and for -1607 2G MMP-1 allele, as well as for -1562 C/T MMP-9
genotype and -1562 T MMP-9 allele in patients with POAG in comparison with
healthy subjects. There was also a positive association of -511 T/T IL-1β genotype as well as -511 T IL-1β allele
occurrence with an increased POAG development risk. Furthermore, an association
of -1607 1G/2G MMP-1, -1562 C/T MMP-9 and -511 C/T IL-1β gene polymorphism with decreased retinal nerve fiber layer thickness in
patients with POAG group was also observed. Results also indicated an
association of 372 T/C TIMP-1 polymorphism with normal range RNFL. Researchers
concluded that -1607 1G/2G MMP-1, -1562 C/T MMP-9, -511 C/T IL-1β SNPs can be considered as important risk factors for POAG[88]. The impact of polymorphic changes in promoter
regions confirmed that allele -1607 1G of MMP-1 gene had 42.91% of -1607 2G
allele transcriptional activity while allele -1562 C of MMP-9 gene showed only
21.86% of -1562 T allele. These results suggest that increased expression
levels of MMPs can be considered as a risk factor for development of POAG[89]. Genetic polymorphism study regarding MMP-9 gene
especially in Caucasian patients confirmed that rs17576 polymorphism is not
related to glaucoma condition but interestingly rs3918249 polymorphism was
found to be a protective factor against glaucoma (Table 1)[90].
Prostaglandin and their analogs commonly used for lowering IOP can
upregulate expression of MMPs-1, 2, 3, 9, and 17, and at the same time can
lower expression levels of TIMP-1 and 2 in human non-pigmented ciliary
epithelial cells[91]. Insights into molecular
mechanisms of segmental aqueous outflow of TM can aid in the design and
delivery of improved treatments for patients suffering from glaucoma because
molecular differences between high and low outflow regions of TM are not
clearly known. An examination of collagen genes such as COL16A1, COL4A2, COL6A1
and 2 and MMP-1, 2, 3 showed enrichment in high flow regions of TM, whereas
COL15A1, and MMP-16 were found to be enriched in low flow regions of TM. These
genes and proteins differences across regions of TM provide evidence for a
molecular basis of segmental flow routes within the aqueous outflow pathway[92]. Additionally, molecular genetic studies have helped
in deciphering the potential causes of disorders in patients with a congenital
optic nerve disease known as cavitary optic disc anomaly (CODA), who are born
with excavation of optic nerve resembling glaucoma. Gene for the
autosomal-dominant CODA was successfully mapped in a large pedigree to a
chromosome 12q locus. Subsequently, in this pedigree a 6-Kbp heterozygous
triplication upstream of MMP-19 gene was discovered. Further characterization
of genetic sequence suggested that triplication of this sequence can lead to
dysregulation of MMP-19 in the CODA patients[93].
It is known that caveolin (CAV) mediated endocytosis process is one of
the mechanisms by which TM cells can control physiological catabolism of ECM to
change the composition of outflow channels inside TM. This is done to regulate
aqueous outflow resistance and dysregulation of CAV functions can contribute to
pathological changes in ECM that are observed in glaucoma patients often. One
SNP was identified between CAV1 and CAV2 on chromosome 7 and was associated
with glaucoma condition. When a CAV-silencing lentiviral vector was employed to
evaluate the effects on ECM turnover by TM cells to measure the effect on
outflow in anterior segment perfusion culture, outflow rates increased
significantly in CAV1-silenced anterior segments, whereas outflow significantly
decreased in CAV2-silenced anterior segments. MMP-2 and MMP-14, and a
disintegrin and metalloproteinase with thrombospondin motifs-4 (ADAMTS4)
colocalized with both CAVs in TM cells. Protein levels and enzyme activities of
MMP/ADAMTS4, fibronectin protein levels, actin stress fibers, and α-smooth muscle actin were all increased in CAV-silenced cells indicating
that dysregulation of CAV functions may contribute to pathological changes in
ECM that are commonly observed in glaucoma patients[94].
Further, abnormal production and accumulation of extracellular
fibrillary material (XFM); a cardinal feature of exfoliation syndrome
represents a pathologic matrix product made by intraocular cells, such as
ciliary epithelial cells, trabecular and corneal endothelial cells,
pre-equatorial epithelial cells of lens, different cell types of iris, as well
as by extraocular cells (vascular cells, fibrocytes, and muscle cells). XFM
composition is not fully known however biochemical analyses have shown a highly
glycosylated, cross-linked, and enzymatically resistant
glycoprotein/proteoglycan complex, composed of a protein core that is
surrounded by glycoconjugates. Protein core includes components of basement
membrane: laminin, nidogen, fibronectin, elastic fiber parts such as
fibrillin-1, elastin, and latent transforming growth factor binding proteins,
as well as enzymes (MMPs), extracellular chaperone clusterin, and cross-linking
enzyme lysyl oxidase-like 1 (LOXL1). LOXL1 is an important cross-linking enzyme
in ECM and is required for the formation of elastic fiber and its
stabilization. It is worth to remember that LOXL1 functions are dysregulated in
glaucoma[95].
Cells involved in exfoliation syndrome exhibit metabolic activation,
such as increased vesicular transport to cell surface, XFM formation within
infoldings of cellular surfaces, and that happens prominently via the
rough ER structures. Also, cells involved in production of XFM display a gene
expression pattern characterized by upregulation of elastic components,
transient upregulation of LOXL1, and dysregulated expression of cytoprotective
gene products, MMPs, and their inhibitors, possibly leading to accumulation and
stable deposition of XFM[96]. Pseudoexfoliation
(PEX) syndrome is a genetically determined disease of ECM and it generally
leads to progressive deposition of fibrillar material in intraocular and
extraocular tissues including TM. It causes open-angle glaucoma (OAG). PEX
process is characterized by an excessive production and abnormal cross-linking
of elastic microfibrils into fibrillar aggregates. Triggering factors include
elevated fibrogenic growth factors (TGF-β1),
reduced proteolytic enzymes, subtle inflammatory processes along with oxidative
stress. Genetic studies identified association between LOXL1 gene polymorphism
with PEX syndrome and glaucoma. PEX familial aggregation suggests genetic
inheritance and has been strongly associated with SNPs of LOXL1 gene on
chromosome 15q24.1. High risk haplotypes vary among different populations but
LOXL1 risk variants occur in almost 100% PEX patients making it a principal
risk factor for PEX phenotype.
DISCUSSION
During past decade, the field of molecular genetics has become one of
the fastest growing disciplines in life sciences heralding development of
potential genomic markers that potentially can identify allelic variants
reproducibly. These tools shall help advance research aiming at diagnosis and treatment
that can ultimately improve clinical ophthalmological practice. Genomic
profiling can assist in understanding genetic determinants of drugs’ response
in patients who could benefit clinically. Certain SNPs such as in PTGFR
(prostaglandin F2α receptor gene) and MMP-1 genes may
determine Latanoprost’s (a commonly used anti-glaucomatous drug) response as
revealed in a study which identified five SNPs related to Latanoprost efficacy.
For example, MMP-1 SNP; rs3753380, has already been associated with a poor
response to Latanoprost in a healthy Japanese population[92].
However, it is too early to derive such parallels from research currently
focusing on epigenetic centered mechanisms operating in a diseased cell.
Epigenetic changes occur without alterations in the actual DNA sequences and
can significantly affect gene transcription in response to environmental
changes.
MMP family of enzymes plays important roles in the physiological and
pathological remodeling of tissues in our body and these set of enzymes achieve
this via an elaborate ECM metabolism[10].
Their dysregulations and subsequent malfunctioning can lead to a variety of eye
diseases because of their crucial roles in many biological processes[7]. Can MMPs and their regulators (TIMPs) be used as potential
markers of ocular diseases in clinics such as for
treating AMD patients soon remains to be seen? There is no cure for the
dry form of AMD and the treatment available for neovascular AMD is via
photodynamic therapy (PDT), or by injections into the vitreous cavities of the
affected eyes that block VEGF actions only slows down the progression of AMD in
a select group of patients. Recent insights into the pathological mechanisms
operating in ECM metabolism may certainly lead to the development of newer and
thus improved therapies for AMD. However, detecting susceptible pool of
patients early on before actual AMD disease symptoms start manifesting
clinically could only be possible by having a robust biomarker tool kit based
on subtle signs of degenerative changes in the photoreceptors and RPE since not
much is known regarding the precise sequence of degenerative changes that
happens during initiation and progression of AMD particularly in the advanced
forms of neovascular AMD phenotype which is preceded by early and intermediate
stages that are characterized by a significant loss of RPE coupled with
associated pigmentary abnormalities[97-100].
Chorio-capillaries, in back of the eyes, are important for survival of
photoreceptors, RPE and adjoining supporting structures. During AMD progression
ECM serves as the area of dynamic changes connected with the activity of its
specific MMPs and TIMPs. Thus, discovery of potential biochemical or genetical
biomarkers based on inflammatory mediators and oxidative stress induced
cellular changes arising from dysregulation of MMPs and TIMPs expression levels
can help identify eye conditions, in advance, in susceptible patient
populations. This can have tremendous value in clinics to predict disease
risks, evaluate treatment efficacies, and to monitor AMD disease progression.
Diabetes has become a medical epidemic of 21st century and is
considered a major public health concern. With over 90% patients with diabetes
it is a tsunami for the risk of developing DR, nephropathy, and neuropathy in affected patients.
Despite ongoing cutting edge research in diabetes field, how exactly retina and
its vasculature are damaged by diabetic milieu
remains quite ambiguous. Environmental factors, life style or disease process
can also bring in modifications in DNA sequence itself, and these modifications
either can silence or activate a specific gene. Diabetic environment
can upregulate or downregulate genes in retina, and latest research shows that
it can also facilitate major epigenetic modifications. Genes associated with
important enzymes such as mitochondrial superoxide dismutase, MMP-9 and
thioredoxin interacting protein and transcriptional factors are epigenetically
modified. Enzymes responsible for these epigenetic modifications are either
activated or inhibited, and levels of microRNAs are also altered[101]. Many metabolic pathways have been implicated in DR
development and thus role of epigenetics in DR is now an emerging area, and recent work has shown
that histone lysine demethylase 1 (LSD1) and DNA methyltransferase are
increased. Thus, a better understanding of these modifications has potential to
identify novel targets to inhibit this devastating disease.
Fortunately, inhibitors and mimics targeted towards histone
modification, DNA methylation, and miRNAs are now being tried for cancer and
other chronic diseases and will open the door for their possible use in
combating diseases such as DR[102].
Additionally, imbalance between MMPs and TIMP (Figure 1) may also promote
neovascularization of retina through PKC activation hence development of novel
compounds with regulatory action on MMPs and TIMPs production through
inhibiting PKC activity can also lead to newer opportunities in developing
therapeutics for treatment and prevention of DR[103]. Further, to
understand better structural basis of TIMPs’ functions and their specificities in vivo, we need to obtain robust
structural results arising from prospective research not only of the strongest
MMP and TIMP complexes but also of full spectrum of PPIs as evidenced by
researchers actively pursuing such ventures. Similarly, regulation of Sirt1
along with pharmaceutical and nutritional means could also serve as a potential
target to prevent or at least delay the development of DR in some patients, if
not in all, because oxidative stress in diabetic individuals inhibits Sirt1
where p65 is hyperacetylated simultaneously increasing binding of p65 at MMP-9
promoter sequence. So, prevention of Sirt1 inhibition via modulating
acetylation of p65 should in principle protect activation of MMP-9 and thus
inhibit DR[104].
Genes that are differentially expressed in glaucoma patients’ ocular
tissue or in cultured human TM cell models are possibly implicated in disease
process and include superoxide dismutase2 (SOD2), aldehyde dehydrogenases 1A1
(ALDH1A1), Microsomal glutathione S-transferase 1 (MGST1), LOX, and LOXL1,
elements of transforming growth factor-β/bone
morphogenetic protein/small body size mothers against decapentaplegic (Smad)
signaling pathways, connective tissue growth factor, MMP-2, TIMP-2, and
endothelin-1 (ET-1). In exfoliation syndrome and XFG fibrillar phenotype, a
proteinaceous extracellular material is produced in excess and accumulates in
both outflow pathways. Material which is locally produced accumulates in the
intertrabecular spaces, juxtacanalicular (JCT) meshwork, and in the inner wall
of Schlemm's canal because of a combination of both excessive synthesis and
insufficient degradation by MMPs. An increase in JCT plaque and decreased
cellularity in TM are thought to contribute to decreased outflow in glaucoma
patients, but XFG patient specimens show reduced extracellular plaque material
in JCT, and the structural integrity of trabecular endothelial cells is mostly
retained and cellularity remains unchanged. Therefore, understanding
distinctions between causes and effects of structural changes that lead to
reduced outflow/elevated IOP are important for developing effective,
individualized treatment strategies in glaucoma[105].
In conclusion, irrespective of their triggers, whether it is oxidative
stress[75,106] or
inflammation[107]. MMPs are capable of degrading
almost all components of ECM and thus play important roles in many physiological
and pathological processes in the eye. On the other hand, TIMPs tend to
neutralize the activities of MMPs and therefore help maintain the stability of
ECM in the ocular compartment. The imbalance between the expression levels of
MMPs and TIMPs has been shown to be closely associated with many ophthalmic
disease conditions (Figure 1). A few were covered in this manuscript however
emerging evidence suggests that MMPs are also associated with pterygium,
corneal lesions and a host of other ocular injuries[108].
Thus, future design and delivery of more targeted, and thereby more effective,
treatments should take into consideration the various molecular genetic aspects
that we have highlighted in our work in this manuscript.
ACKNOWLEDGEMENTS
We
sincerely thank Aman Babbarwal and Karan Babbarwal for their excellent editing
skills and meaningful inputs in this manuscript.
Foundations:
Supported in part by NIH Heart, Lung, and Blood Institute
(No.HLO74815); Institute of Neurological Disorders and Stroke (No.NS-084823).
Conflicts
of Interest: Singh M, None; Tyagi SC, None.
REFERENCES
1 Stamenkovic I. Extracellular matrix remodelling: the
role of matrix metalloproteinases. J
Pathol 2003;200(4):448-464. [CrossRef] [PubMed]
2 Parks WC, Wilson CL, Lopez-Boado YS. Matrix
metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 2004;4(8):617-629. [CrossRef] [PubMed]
3 Van Wart HE, Birkedal-Hansen H. The cysteine switch:
a principle of regulation of metalloproteinase activity with potential
applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci U S A
1990;87(14):5578-5582. [CrossRef]
4 Rosenblum G, Van den Steen PE, Cohen SR, Grossmann
JG, Frenkel J, Sertchook R, Slack N, Strange RW, Opdenakker G, Sagi I. Insights
into the structure and domain flexibility of full-length pro-matrix
metalloproteinase-9/gelatinase B. Structure
2007;15(10):1227-1236. [CrossRef] [PubMed]
5 Brew K, Nagase H. The tissue inhibitors of
metalloproteinases (TIMPs): an ancient family with structural and functional
diversity. Biochim Biophys Acta
2010;1803(1):55-71. [CrossRef] [PMC free article] [PubMed]
6 Murphy G. Tissue inhibitors of metalloproteinases. Genome Biol 2011;12(11):233. [CrossRef] [PMC free article] [PubMed]
7 Itoh Y, Nagase H. Matrix metalloproteinases in
cancer. Essays Biochem 2002;38:21-36.
[CrossRef] [PubMed]
8 Abu El-Asrar AM, Mohammad G, Nawaz MI, Siddiquei MM,
Van den Eynde K, Mousa A, De Hertogh G, Opdenakker G. Relationship between
vitreous levels of matrix metalloproteinases and vascular endothelial growth
factor in proliferative diabetic retinopathy. PLoS One 2013;8(12):e85857. [CrossRef] [PMC free article] [PubMed]
9 Murphy G, Nagase H. Progress in matrix
metalloproteinase research. Mol Aspects
Med 2008;29(5):290-308. [CrossRef] [PMC free article] [PubMed]
11 Westermarck J, Kahari VM. Regulation of matrix
metalloproteinase expression in tumor invasion. FASEB J 1999;13(8):781-792. [PubMed]
12 Nicholson R, Murphy G, Breathnach R. Human and rat
malignant-tumor-associated mRNAs encode stromelysin-like metalloproteinases. Biochemistry 1989;28(12):5195-5203. [CrossRef] [PubMed]
13 Nakamura H, Fujii Y, Ohuchi E, Yamamoto E, Okada Y.
Activation of the precursor of human stromelysin 2 and its interactions with
other matrix metalloproteinases. Eur J
Biochem 1998;253(1):67-75. [CrossRef] [PubMed]
14 Brown D, Hamdi H, Bahri S, Kenney MC.
Characterization of an endogenous metalloproteinase in human vitreous. Curr Eye Res 1994;13(9):639-647. [CrossRef]
15 Plantner JJ, Smine A, Quinn TA. Matrix
metalloproteinases and metalloproteinase inhibitors in human interphotoreceptor
matrix and vitreous. Curr Eye Res
1998;17(2):132-140. [CrossRef]
16 Kawashima Y, Saika S, Miyamoto T, Yamanaka O, Okada
Y, Tanaka S, Ohnishi Y. Matrix metalloproteinases and tissue inhibitors of
metalloproteinases of fibrous humans lens capsules with intraocular lenses. Curr Eye Res 2000;21(6):962-967. [CrossRef]
17 Daniels JT, Geerling G, Alexander RA, Murphy G,
Khaw PT, Saarialho-Kere U. Temporal and spatial expression of matrix metalloproteinases
during wound healing of human corneal tissue. Exp Eye Res 2003;77(6):653-664. [CrossRef]
18 Weber KT, Sun Y, Tyagi SC, Cleutjens JP. Collagen
network of the myocardium: function, structural remodeling and regulatory
mechanisms. J Mol Cell Cardiol
1994;26(3):279-292. [CrossRef] [PubMed]
19 Tyagi SC, Kumar SG, Haas SJ, Reddy HK, Voelker DJ,
Hayden MR, Demmy TL, Schmaltz RA, Curtis JJ. Post-transcriptional regulation of
extracellular matrix metalloproteinase in human heart end-stage failure
secondary to ischemic cardiomyopathy. J
Mol Cell Cardiol 1996;28(7):1415-1428. [CrossRef] [PubMed]
20 Tyagi SC, Joshua IG. Exercise and nutrition in
myocardial matrix metabolism, remodeling, regeneration, epigenetics, microcirculation,
and muscle. Can J Physiol Pharmacol
2014;92(7):521-523. [CrossRef] [PubMed]
21 Vacek TP, Rehman S, Neamtu D, Yu S, Givimani S, Tyagi
SC. Matrix metalloproteinases in atherosclerosis: role of nitric oxide,
hydrogen sulfide, homocysteine, and polymorphisms. Vasc Health Risk Manag 2015;11:173-183. [CrossRef] [PMC free article] [PubMed]
22 Givvimani S, Kundu S, Narayanan N, Armaghan F,
Qipshidze N, Pushpakumar S, Vacek TP, Tyagi SC. TIMP-2 mutant decreases MMP-2
activity and augments pressure overload induced LV dysfunction and heart
failure. Arch Physiol Biochem
2013;119(2):65-74. [CrossRef] [PMC free article] [PubMed]
23 Lominadze D, Roberts AM, Tyagi N, Moshal KS, Tyagi
SC. Homocysteine causes cerebrovascular leakage in mice. Am J Physiol Heart Circ Physiol 2006;290(3):H1206-H1213. [CrossRef] [PMC free article] [PubMed]
24 Amin M, Pushpakumar S, Muradashvili N, Kundu S,
Tyagi SC, Sen U. Regulation and involvement of matrix metalloproteinases in
vascular diseases. Front Biosci (Landmark
Ed) 2016;21:89-118. [CrossRef]
25 Mishra PK, Tyagi N, Sen U, Joshua IG, Tyagi SC.
Synergism in hyperhomocysteinemia and diabetes: role of PPAR gamma and tempol. Cardiovasc Diabetol 2010;9:49. [CrossRef] [PMC free article] [PubMed]
26 Sen U, Rodriguez WE, Tyagi N, Kumar M, Kundu S,
Tyagi SC. Ciglitazone, a PPARgamma agonist, ameliorates diabetic nephropathy in
part through homocysteine clearance. Am J
Physiol Endocrinol Metab 2008;295(5):E1205-E1212. [CrossRef] [PMC free article] [PubMed]
28 Givvimani S, Munjal C, Tyagi N, Sen U, Metreveli N,
Tyagi SC. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates
pressure overload induced heart failure. PLoS
One 2012;7(3):e32388. [CrossRef] [PMC free article] [PubMed]
29 Rawlings ND, Barrett AJ, Bateman A. MEROPS: the
database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 2012;40(Database
issue):D343-D350. [CrossRef] [PMC free article] [PubMed]
30 Kveiborg M, Jacobsen J, Lee MH, Nagase H, Wewer UM,
Murphy G. Selective inhibition of ADAM12 catalytic activity through engineering
of tissue inhibitor of metalloproteinase 2 (TIMP-2). Biochem J 2010;430(1): 79-86. [CrossRef] [PMC free article] [PubMed]
31 Hutton M, Willenbrock F, Brocklehurst K, Murphy G.
Kinetic analysis of the mechanism of interaction of full-length TIMP-2 and
gelatinase A: evidence for the existence of a low-affinity intermediate. Biochemistry 1998;37(28):10094-10098. [CrossRef] [PubMed]
32 Batra J, Robinson J, Soares AS, Fields AP, Radisky
DC, Radisky ES. Matrix metalloproteinase-10 (MMP-10) interaction with tissue
inhibitors of metalloproteinases TIMP-1 and TIMP-2: binding studies and crystal
structure. J Biol Chem
2012;287(19):15935-15946. [CrossRef] [PMC free article] [PubMed]
33 Stetler-Stevenson WG. The tumor microenvironment:
regulation by MMP-independent effects of tissue inhibitor of
metalloproteinases-2. Cancer Metastasis
Rev 2008;27(1):57-66. [CrossRef] [PMC free article] [PubMed]
34 Gomis-Ruth FX, Maskos K, Betz M, Bergner A, Huber R,
Suzuki K, Yoshida N, Nagase H, Brew K, Bourenkov GP, Bartunik H, Bode W.
Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by
TIMP-1. Nature 1997;389(6646):77-81.
[CrossRef] [PubMed]
35 Fernandez-Catalan C, Bode W, Huber R, Turk D,
Calvete JJ, Lichte A, Tschesche H, Maskos K. Crystal structure of the complex
formed by the membrane type 1-matrix metalloproteinase with the tissue
inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. EMBO J 1998;17(17):5238-5248. [CrossRef] [PMC free article] [PubMed]
36 Maskos K, Lang R, Tschesche H, Bode W. Flexibility
and variability of TIMP binding: X-ray structure of the complex between
collagenase-3/MMP-13 and TIMP-2. J Mol
Biol 2007;366(4):1222-1231. [CrossRef] [PubMed]
37 Grossman M, Tworowski D, Dym O, Lee MH, Levy Y,
Murphy G, Sagi I. The intrinsic protein flexibility of endogenous protease
inhibitor TIMP-1 controls its binding interface and affects its function. Biochemistry 2010;49(29):6184-6192. [CrossRef] [PubMed]
38 Iyer S, Wei S, Brew K, Acharya KR. Crystal
structure of the catalytic domain of matrix metalloproteinase-1 in complex with
the inhibitory domain of tissue inhibitor of metalloproteinase-1. J Biol Chem 2007;282(1):364-371. [CrossRef] [PubMed]
39 Piso DY, Ribeiro AP, Silva ML, Guimaraes PJ,
Morales A, Martins BC, Padua IM, Renzo R, Andrade AL, Uscategui RR, Laus JL.
Effects of antiproteolytic agents on corneal epithelial viability and matrix
metalloproteinase-2 and metalloproteinase-9 activity in alkali-burned corneas
of rats. Vet Ophthalmol
2014;17(1):23-31. [CrossRef] [PubMed]
40 Matsubara M, Girard MT, Kublin CL, Cintron C, Fini
ME. Differential roles for two gelatinolytic enzymes of the matrix
metalloproteinase family in the remodelling cornea. Dev Biol 1991;147(2):425-439. [CrossRef]
42 Kutty RK, Samuel W, Boyce K, Cherukuri A, Duncan T,
Jaworski C, Nagineni CN, Redmond TM. Proinflammatory cytokines decrease the
expression of genes critical for RPE function. Mol Vis 2016;22:1156-1168. [PMC free article] [PubMed]
43 Friedman DS, O'Colmain BJ, Munoz B, Tomany SC,
McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J. Prevalence of
age-related macular degeneration in the United States. Arch Ophthalmol 2004;122(4): 564-572. [CrossRef] [PubMed]
44 Sobrin L, Seddon JM. Nature and nurture- genes and
environment- predict onset and progression of macular degeneration. Prog Retin Eye Res 2014;40:1-15. [CrossRef] [PubMed]
45 Seddon JM, McLeod DS, Bhutto IA, Villalonga MB,
Silver RE, Wenick AS, Edwards MM, Lutty GA. Histopathological insights into choroidal
vascular loss in clinically documented cases of age-related macular
degeneration. JAMA Ophthalmol
2016;134(11):1272-1280. [CrossRef] [PubMed]
46 Fritsche LG, Chen W, Schu M, et al. Seven new loci associated with age-related macular
degeneration. Nat Genet
2013;45(4):433-439. [CrossRef] [PMC free article] [PubMed]
47 Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY.
Age-related macular degeneration. Lancet
2012;379(9827):1728-1738. [CrossRef]
48 Fiotti N, Pedio M, Battaglia Parodi M, Altamura N,
Uxa L, Guarnieri G, Giansante C, Ravalico G. MMP-9 microsatellite polymorphism
and susceptibility to exudative form of age-related macular degeneration. Genet Med 2005;7(4):272-277. [CrossRef]
49 Langton KP, McKie N, Curtis A, Goodship JA, Bond
PM, Barker MD, Clarke M. A novel tissue inhibitor of metalloproteinases-3
mutation reveals a common molecular phenotype in Sorsby's fundus dystrophy. J Biol Chem 2000;275(35):27027-27031. [PubMed]
50 Liutkeviciene R, Lesauskaite V, Sinkunaite-Marsalkiene
G, Zaliuniene D, Zaliaduonyte-Peksiene D, Mizariene V, Gustiene O, Jasinskas V,
Jariene G, Tamosiunas A. The role of matrix metalloproteinases polymorphisms in
age-related macular degeneration. Ophthalmic
Genet 2015;36(2):149-155. [CrossRef] [PubMed]
51 Ortak H, Demir S, Ates O, Benli I, Sogut E, Sahin
M. The role of MMP2 (-1306C>T) and TIMP2 (-418 G>C) promoter variants in
age-related macular degeneration. Ophthalmic
Genet 2013;34(4):217-222. [CrossRef] [PubMed]
52 Seitzman RL, Mahajan VB, Mangione C, Cauley JA,
Ensrud KE, Stone KL, Cummings SR, Hochberg MC, Hillier TA, Sinsheimer JS, Yu F,
Coleman AL. Estrogen receptor alpha and matrix metalloproteinase 2
polymorphisms and age-related maculopathy in older women. Am J Epidemiol 2008;167(10):1217-1225. [CrossRef] [PubMed]
53 Weber BH, Vogt G, Pruett RC, Stohr H, Felbor U.
Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with
Sorsby's fundus dystrophy. Nat Genet
1994;8(4):352-356. [CrossRef] [PubMed]
54 Raffetto JD, Khalil RA. Matrix metalloproteinases
and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol 2008; 75(2):346-359. [CrossRef] [PMC free article] [PubMed]
55 Chen W, Stambolian D, Edwards AO, et al. Genetic variants near TIMP3 and
high-density lipoprotein-associated loci influence susceptibility to
age-related macular degeneration. Proc
Natl Acad Sci U S A 2010; 107(16):7401-7406. [CrossRef] [PMC free article] [PubMed]
56 Fariss RN, Apte SS, Olsen BR, Iwata K, Milam AH.
Tissue inhibitor of metalloproteinases-3 is a component of Bruch's membrane of
the eye. Am J Pathol
1997;150(1):323-328. [PMC free article] [PubMed]
57 Kamei M, Hollyfield JG. TIMP-3 in Bruch's membrane:
changes during aging and in age-related macular degeneration. Invest Ophthalmol Vis Sci
1999;40(10):2367-2375. [PubMed]
58 Leu ST, Batni S, Radeke MJ, Johnson LV, Anderson
DH, Clegg DO. Drusen are cold spots for proteolysis: expression of matrix
metalloproteinases and their tissue inhibitor proteins in age-related macular
degeneration. Exp Eye Res
2002;74(1):141-154. [CrossRef] [PubMed]
59 Chau KY, Sivaprasad S, Patel N, Donaldson TA, Luthert
PJ, Chong NV. Plasma levels of matrix metalloproteinase-2 and -9 (MMP-2 and
MMP-9) in age-related macular degeneration. Eye
(Lond) 2008;22(6):855-859. [CrossRef] [PubMed]
60 Zeng R, Wen F, Zhang X, Su Y. Serum levels of
matrix metalloproteinase 2 and matrix metalloproteinase 9 elevated in
polypoidal choroidal vasculopathy but not in age-related macular degeneration. Mol Vis 2013;19:729-736. [PMC free article] [PubMed]
61 Jin E, Bai Y, Huang L, Zhao M, Zhang C, Zhao M, Li
X. Evidence of a novel gene HERPUD1 in polypoidal choroidal vasculopathy. Int J Clin Exp Pathol
2015;8(11):13928-13944. [PMC free article] [PubMed]
62 Yannuzzi LA, Ciardella A, Spaide RF, Rabb M, Freund
KB, Orlock DA. The expanding clinical spectrum of idiopathic polypoidal
choroidal vasculopathy. Arch Ophthalmol
1997;115(4):478-485. [CrossRef]
63 Plantner JJ, Jiang C, Smine A. Increase in
interphotoreceptor matrix gelatinase A (MMP-2) associated with age-related
macular degeneration. Exp Eye Res
1998;67(6):637-645. [CrossRef] [PubMed]
64 Hussain AA, Lee Y, Zhang JJ, Marshall J. Disturbed
matrix metalloproteinase activity of Bruch's membrane in age-related macular
degeneration. Invest Ophthalmol Vis Sci
2011;52(7):4459-4466. [CrossRef] [PubMed]
65 Ahir A, Guo L, Hussain AA, Marshall J. Expression
of metalloproteinases from human retinal pigment epithelial cells and their
effects on the hydraulic conductivity of Bruch's membrane. Invest Ophthalmol Vis Sci 2002;43(2):458-465. [PubMed]
66 Roider J, Hillenkamp F, Flotte T, Birngruber R.
Microphotocoagulation: selective effects of repetitive short laser pulses. Proc Natl Acad Sci U S A
1993;90(18):8643-8647. [CrossRef]
67 Zhang JJ, Sun Y, Hussain AA, Marshall J. Laser-mediated
activation of human retinal pigment epithelial cells and concomitant release of
matrix metalloproteinases. Invest
Ophthalmol Vis Sci 2012;53(6):2928-2937. [CrossRef] [PubMed]
68 Brownlee M. The pathobiology of diabetic
complications: a unifying mechanism. Diabetes
2005;54(6):1615-1625. [CrossRef]
69 Petrovic D. Candidate genes for proliferative
diabetic retinopathy. Biomed Res Int
2013;2013:540416. [CrossRef] [PMC free article] [PubMed]
70 Beranek M, Kolar P, Tschoplova S, Kankova K, Vasku
A. Genetic variations and plasma levels of gelatinase A (matrix
metalloproteinase-2) and gelatinase B (matrix metalloproteinase-9) in
proliferative diabetic retinopathy. Mol
Vis 2008;14:1114-1121. [PMC free article] [PubMed]
71 Maeda S, Haneda M, Guo B, Koya D, Hayashi K,
Sugimoto T, Isshiki K, Yasuda H, Kashiwagi A, Kikkawa R. Dinucleotide repeat
polymorphism of matrix metalloproteinase-9 gene is associated with diabetic
nephropathy. Kidney Int
2001;60(4):1428-1434. [CrossRef] [PubMed]
73 Yang J, Fan XH, Guan YQ, Li Y, Sun W, Yang XZ, Liu R.
MMP-2 gene polymorphisms in type 2 diabetes mellitus diabetic retinopathy. Int J Ophthalmol 2010;3(2):137-140. [PMC free article] [PubMed]
74 Reddy MA, Natarajan R. Epigenetic mechanisms in
diabetic vascular complications. Cardiovasc
Res 2011;90(3):421-429. [CrossRef] [PMC free article] [PubMed]
75 Singh M, Kapoor A, Bhatnagar A. Oxidative and
reductive metabolism of lipid-peroxidation derived carbonyls. Chem Biol Interact 2015;234:261-273. [CrossRef] [PMC free article] [PubMed]
76 Kowluru RA, Kanwar M. Oxidative stress and the
development of diabetic retinopathy: contributory role of matrix
metalloproteinase-2. Free Radic Biol Med
2009;46(12):1677-1685. [CrossRef] [PMC free article] [PubMed]
77 Giebel SJ, Menicucci G, McGuire PG, Das A. Matrix
metalloproteinases in early diabetic retinopathy and their role in alteration
of the blood-retinal barrier. Lab Invest
2005;85(5):597-607. [CrossRef] [PubMed]
78 Descamps FJ, Martens E, Kangave D, Struyf S, Geboes
K, Van Damme J, Opdenakker G, Abu El-Asrar AM. The activated form of gelatinase
B/matrix metalloproteinase-9 is associated with diabetic vitreous hemorrhage. Exp Eye Res 2006;83(2):401-407. [CrossRef] [PubMed]
79 Mohammad G, Kowluru RA. Diabetic retinopathy and
signaling mechanism for activation of matrix metalloproteinase-9. J Cell Physiol 2012;227(3):1052-1061. [CrossRef] [PMC free article] [PubMed]
80 Zhong Q, Kowluru RA. Regulation of matrix
metalloproteinase-9 by epigenetic modifications and the development of diabetic
retinopathy. Diabetes
2013;62(7):2559-2568. [CrossRef] [PMC free article] [PubMed]
81 Kosano H, Okano T, Katsura Y, Noritake M, Kado S,
Matsuoka T, Nishigori H. ProMMP-9 (92 kDa gelatinase) in vitreous fluid of patients
with proliferative diabetic retinopathy. Life
Sci 1999;64(25):2307-2315. [CrossRef]
82 Salzmann J, Limb GA, Khaw PT, Gregor ZJ, Webster L,
Chignell AH, Charteris DG. Matrix metalloproteinases and their natural
inhibitors in fibrovascular membranes of proliferative diabetic retinopathy. Br J Ophthalmol 2000;84(10):1091-1096. [CrossRef]
83 Saghizadeh M, Brown DJ, Castellon R, et al. Overexpression of matrix
metalloproteinase-10 and matrix metalloproteinase-3 in human diabetic corneas:
a possible mechanism of basement membrane and integrin alterations. Am J Pathol 2001;158(2):723-734. [CrossRef]
84 Jin M, Kashiwagi K, Iizuka Y, Tanaka Y, Imai M,
Tsukahara S. Matrix metalloproteinases in human diabetic and nondiabetic
vitreous. Retina 2001;21(1):28-33. [CrossRef]
85 Noda K, Ishida S, Inoue M, Obata K, Oguchi Y, Okada
Y, Ikeda E. Production and activation of matrix metalloproteinase-2 in
proliferative diabetic retinopathy. Invest
Ophthalmol Vis Sci 2003;44(5):2163-2170. [CrossRef]
86 Konieczka K, Frankl S, Todorova MG, Henrich PB.
Unstable oxygen supply and glaucoma. Klin
Monbl Augenheilkd 2014;231(2):121-126. [CrossRef] [PubMed]
87 Ashworth Briggs EL, Toh T, Eri R, Hewitt AW, Cook
AL. TIMP1, TIMP2, and TIMP4 are increased in aqueous humor from primary open
angle glaucoma patients. Mol Vis
2015;21:1162-1172. [PubMed]
88 Markiewicz L, Majsterek I, Przybylowska K, Dziki L,
Waszczyk M, Gacek M, Kaminska A, Szaflik J, Szaflik JP. Gene polymorphisms of
the MMP1, MMP9, MMP12, IL-1beta and TIMP1 and the risk of primary open-angle
glaucoma. Acta Ophthalmol
2013;91(7):e516-e523. [CrossRef] [PubMed]
89 Markiewicz L, Pytel D, Mucha B, Szymanek K, Szaflik
J, Szaflik JP, Majsterek I. Altered expression levels of MMP1, MMP9, MMP12,
TIMP1, and IL-1beta as a risk factor for the elevated IOP and optic nerve head
damage in the primary open-angle glaucoma patients. Biomed Res Int 2015;2015:812503. [CrossRef] [PMC free article] [PubMed]
90 Zhang Y, Wang M, Zhang S. Association of MMP-9 gene
polymorphisms with glaucoma: a Meta-analysis. Ophthalmic Res 2016;55(4):172-179. [CrossRef] [PubMed]
91 Yamada H, Yoneda M, Gosho M, Kato T, Zako M.
Bimatoprost, latanoprost, and tafluprost induce differential expression of
matrix metalloproteinases and tissue inhibitor of metalloproteinases. BMC Ophthalmol 2016;16:26. [CrossRef] [PMC free article] [PubMed]
92 Vranka JA, Bradley JM, Yang YF, Keller KE, Acott
TS. Mapping molecular differences and extracellular matrix gene expression in
segmental outflow pathways of the human ocular trabecular meshwork. PLoS One 2015;10(3):e0122483. [CrossRef] [PMC free article] [PubMed]
93 Hazlewood RJ, Roos BR, Solivan-Timpe F, Honkanen
RA, Jampol LM, Gieser SC, Meyer KJ, Mullins RF, Kuehn MH, Scheetz TE, Kwon YH,
Alward WL, Stone EM, Fingert JH. Heterozygous triplication of upstream
regulatory sequences leads to dysregulation of matrix metalloproteinase 19 in
patients with cavitary optic disc anomaly. Hum
Mutat 2015;36(3):369-378. [CrossRef] [PMC free article] [PubMed]
94 Aga M, Bradley JM, Wanchu R, Yang YF, Acott TS,
Keller KE. Differential effects of caveolin-1 and -2 knockdown on aqueous
outflow and altered extracellular matrix turnover in caveolin-silenced
trabecular meshwork cells. Invest
Ophthalmol Vis Sci 2014;55(9):5497-5509. [CrossRef] [PMC free article] [PubMed]
95 Elhawy E, Kamthan G, Dong CQ, Danias J.
Pseudoexfoliation syndrome, a systemic disorder with ocular manifestations. Hum Genomics 2012;6:22. [CrossRef] [PMC free article] [PubMed]
96 Zenkel M, Schlotzer-Schrehardt U. The composition
of exfoliation material and the cells involved in its production. J Glaucoma 2014;23(8 Suppl 1):S12-S14. [CrossRef] [PubMed]
97 McLeod DS, Taomoto M, Otsuji T, Green WR, Sunness
JS, Lutty GA. Quantifying changes in RPE and choroidal vasculature in eyes with
age-related macular degeneration. Invest
Ophthalmol Vis Sci 2002;43(6): 1986-1993. [PubMed]
98 McLeod DS, Grebe R, Bhutto I, Merges C, Baba T,
Lutty GA. Relationship between RPE and choriocapillaris in age-related macular
degeneration. Invest Ophthalmol Vis Sci
2009;50(10):4982-4991. [CrossRef] [PMC free article] [PubMed]
99 Bhutto I, Lutty G. Understanding age-related
macular degeneration (AMD): relationships between the photoreceptor/retinal
pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol Aspects Med 2012;33(4):295-317. [CrossRef] [PMC free article] [PubMed]
100 Biesemeier A, Taubitz T, Julien S, Yoeruek E,
Schraermeyer U. Choriocapillaris breakdown precedes retinal degeneration in
age-related macular degeneration. Neurobiol
Aging 2014;35(11):2562-2573. [CrossRef] [PubMed]
101 Kowluru RA, Mishra M. Contribution of epigenetics
in diabetic retinopathy. Sci China Life
Sci 2015;58(6):556-563. [CrossRef] [PubMed]
102 Kowluru RA, Santos JM, Mishra M. Epigenetic
modifications and diabetic retinopathy. Biomed
Res Int 2013;2013:635284. [CrossRef]
103 Miyata Y, Kase M, Sugita Y, Shimada A, Nagase T, Katsura
Y, Kosano H. Protein kinase C-mediated regulation of matrix metalloproteinase
and tissue inhibitor of metalloproteinase production in a human retinal müller
cells. Curr Eye Res
2012;37(9):842-849. [CrossRef] [PubMed]
104 Kowluru RA, Santos JM, Zhong Q. Sirt1, a negative
regulator of matrix metalloproteinase-9 in diabetic retinopathy. Invest Ophthalmol Vis Sci
2014;55(9):5653-5660. [CrossRef] [PMC free article] [PubMed]
105 Rasmussen CA, Kaufman PL. The trabecular meshwork
in normal eyes and in exfoliation glaucoma. J
Glaucoma 2014;23(8 Suppl 1): S15-S19. [CrossRef] [PMC free article] [PubMed]
106 Gencer S, Cebeci A, Irmak-Yazicioglu MB. Matrix
metalloproteinase gene expressions might be oxidative stress targets in gastric
cancer cell lines. Chin J Cancer Res
2013;25(3):322-333. [PMC free article] [PubMed]
107 Singh M, Tyagi SC. Homocysteine mediates
transcriptional changes of the inflammatory pathway signature genes in human
retinal pigment epithelial cells. Int J
Ophthalmol 2017;10(5):696-704. [PMC free article] [PubMed]
108 Xiang MH, Zhang XR, Zhang ZY, Li QS, Wang HM, Han
ZM, Zhou HM, Jia YL, Chen XX. Expressions of matrix metalloproteinases 1 and 3
and their tissue inhibitors in the conjunctival tissue and fibroblasts cultured
from conjunctivochalasis. Int J Ophthalmol
2017;10(4): 555-559. [PMC free article] [PubMed]