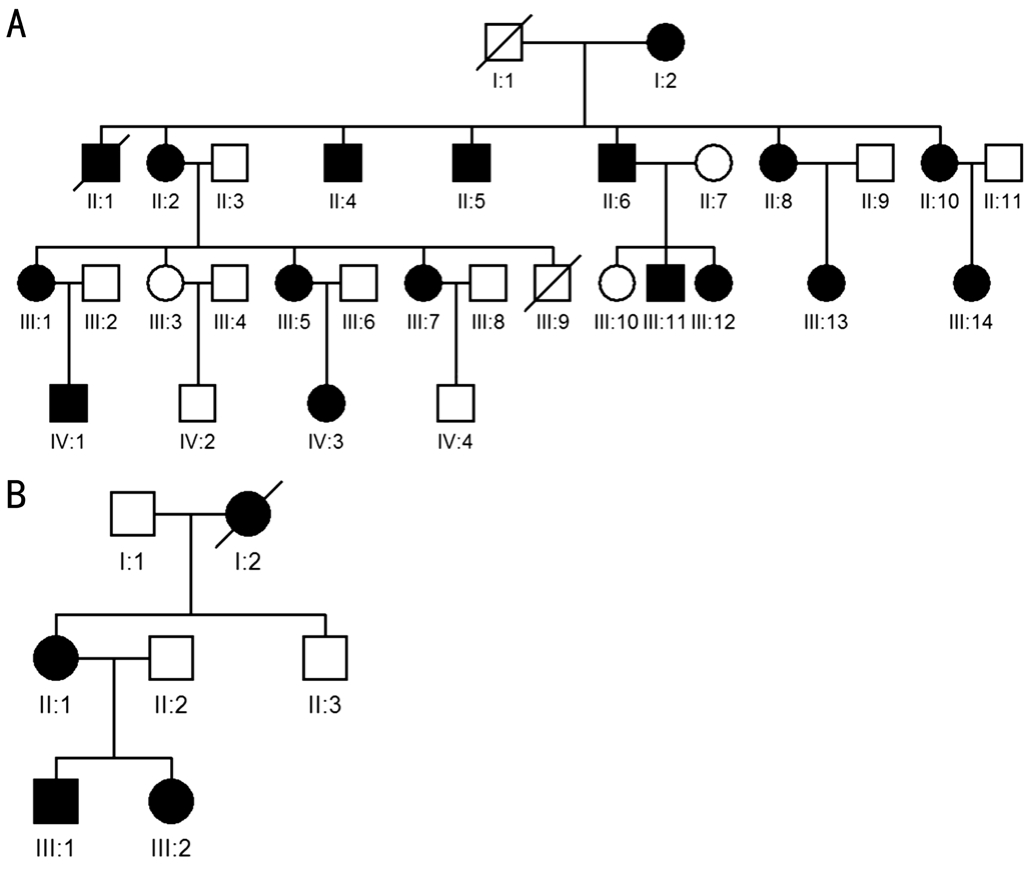
Figure 1 Pedigrees of two Chinese families with congenital cataract
Filled square: Male patient; Filled circle: Female patient;Empty symbol: Normal healthy individual; “/”: Deceased person.
·Basic Research·
Peng Chen1, Hao Chen1, Xiao-Jing Pan2, Su-Zhen Tang1, Yu-Jun Xia1, Hui Zhang3
1Qingdao University, Qingdao 266071, Shandong Province,China
2Shandong Eye Institute, Qingdao 266071, Shandong Province,China
3Jinan Second People’s Hospital, Jinan 250022, Shandong Province, China
Abstract
●AlM:To summarize the phenotypes and identify the underlying genetic cause of theCRYBB1andCRYBB2gene responsible for congenital cataract in two Chinese families.
●METHODS:Detailed family histories and clinical data were collected from patients during an ophthalmologic examination. Of 523 inheritable genetic vision systemrelated genes were captured and sequenced by targeted next-generation sequencing, and the results were confirmed by Sanger sequencing. The possible functional impacts of an amino acid substitution were performed with PolyPhen-2 and SlFT predictions.
●RESULTS:The patients in the two families were affected with congenital cataract. Sixty-five (FAMlLY-1) and sixtytwo (FAMlLY-2) single-nucleotide polymorphisms and indels were selected by recommended filtering criteria.Segregation was then analyzed by applying Sanger sequencing with the family members. A heterozygousCRYBB1mutation in exon 4 (c.347T>C, p.L116P) was identified in sixteen patients in FAMlLY-1. A heterozygousCRYBB2mutation in exon 5 (c.355G>A, p.G119R) was identified in three patients in FAMlLY-2. Each mutation cosegregated with the affected individuals and did not exist in unaffected family members and 200 unrelated normal controls.The mutation was predicted to be highly conservative and to be deleterious by both PolyPhen-2 and SlFT.
●CONCLUSlON:TheCRYBB1mutation (c.347T>C)andCRYBB2mutation (c.355G>A) are novel in patients with congenital cataract. We summarize the variable phenotypes among the patients, which expanded the phenotypic spectrum of congenital cataract in a different ethnic background.
●KEYWORDS:CRYBB1;CRYBB2; next-generation sequencing; congenital cataract
The majority of blindness cases in children result from congenital cataract (OMIM 601547). Congenital cataract refers to lens opacity that occurs in the first year of life, with an incidence of about 1/10 000 to 3/10 000[1]. Worldwide,more than 1 million children are blind because of cataracts[2].
Congenital cataracts occur independently or as part of multisystem abnormalities. Its most common pattern is autosomal dominance, but other genetic patterns have been reported[3].
Previous studies have reported a total of more than 200 genes or loci associated with cataract (Cat-Map)[4]. There are about 45 chromosome loci and 38 genes associated with non-syndromic cataracts[5]. About half of all inherited cataracts are caused by mutations in crystallins[3]. The order and stability of crystallin are key factors in maintaining lens transparency[6]. Some mutations in crystallin cause aggregation of mutant proteins that lead to congenital cataracts, which are usually monogenic diseases consistent with the Mendelian pattern. Other mutations, which increase the susceptibility of individuals to the environment, often contribute to the development of age-related cataracts, which are usually multifactorial[3].
Other pathogenic genes of cataract have been reported,e.g.lens specific connexins, major intrinsic protein, paired-like homeodomain transcription factor-3, aquaporin-0, lens intrinsic membrane protein 2, avian musculo-aponeurotic fibrosarcoma,and beaded filament structural proteins-2[6-13].
Compared with traditional approaches including direct sequencing and linkage disequilibrium, next-generation sequencing (NGS) has been demonstrated as a significant improvement that provides precise diagnostic information and extends the possibility of targeted treatments. In this study, a gene capture panel was used to encompass the exons and untranslated region of 523 genes related to inherited eye disorders. The capture probes were custom designed and produced by Joy Orient Translational Medicine Research(Beijing, China). Two new heterozygousmutations were identified in two congenital cataract families in China,including c.347T>C [p.L116P; exon 4 ofCRYBB1,with cosegregation in 65 single-nucleotide polymorphisms (SNPs;FAMILY-1)] and c.355G>A [p.G119R; exon 5 ofCRYBB2,with co-segregation in 62 SNPs (FAMILY-2)]. By functional prediction, we found that these two missense mutations were pathogenic, and may be the main cause of cataract formation in the two families. Our results demonstrated that the gene testing panel is a cost-effective and high-throughput method that could be applied in both research and clinically molecular diagnosis of genetic eye diseases.
Subject Recruitment and Clinical ExaminationTwo families of autosomal dominant congenital nuclear cataracts were recruited at the Second People’s Hospital of Jinan, China.Both families were from Linyi (Shandong, China). There were 28 people in FAMILY-1 (16 affected, 12 unaffected; 14 males and 14 females). There were 6 people in FAMILY-2 (3 affected, 3 unaffected; 4 males and 2 females). Members of both families had no other system anomalies. The study was conducted in accordance with the Helsinki Declaration and received informed consent from all participants. Diagnosis was confirmed by ophthalmologic examinations, including visual acuity, slit lamp examination, tonometer, corneal curvature measurement, corneal endothelium examination, ultrasound A/B scan or cataract extraction history. Eye photos were taken by slit lamp photography without pupil dilation. Totally 200 subjects from the same population without diagnostic features of congenital cataract were recruited as normal controls
Targeted Next-generation SequencingOf 5 mL of venous blood was taken from each participant, and genomic DNA was extracted using a DNA isolation kit for mammalian blood(Tiangen, Beijing, China). Of 523 genetic vision system related genes were respectively sequenced for II:5 of FAMILY-1 and II:1 of FAMILY-2 (Joy orient translational medicine research,Beijing, China). Burrows Wheeler Aligner software was used to perform short read mapping and alignment. SOAPsnp software and GATK Indel Genotyper were used to test SNPs and insertions/deletions respectively.
Variant AnalysisData was provided as lists of sequence variants (SNPs and short indels), relative to the reference genome. Identified variants were filtered against the Single Nucleotide Polymorphism Database (dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi/), 1000 Genomes Project (http://www.1000genome.org/), HapMap 8 (http://hapmap.ncbi.nlm.nih.gov/) database, and YH database[14].
Verification of VariantsSanger sequencing was carried out in two siblings and their children (II:2, II:6, III:3, III:7, III:10,III:12) to conform whether there were variants may participate in the disease process in FAMILY-1. DNA sequencing was further performed to screen theCRYBB1gene in all individuals in FAMILY-1. Sanger sequencing was also performed on all individuals to verify whether the potential candidate variants detected by targeted NGS were co-segregated with the disease phenotype in FAMILY-2.
Primers for flanking candidate loci were designed and synthesized. Each target fragment was amplified using Taq DNA polymerase (Takara, Dalian, China). Direct DNA sequencing was performed using the BigDyeTM Terminator v3.1 Cycle Sequencing Kit and the ABI PRISM 3730 Sequencer(Applied Biosystems Inc., USA). DNASTAR software package (DNASTAR Inc., USA) was used to align and analyze the sequences. The novel mutations ofCRYBB1/CRYBB2were also genotyped in 200 unrelated healthy controls Sanger sequencing. The Sanger sequencing method is the same as the above description.
Clinical Assessment and FindingsIn this study, there were two Chinese pedigrees with an autosomal dominant inheritance pattern, both of which had congenital nuclear cataracts(Figure 1). FAMILY-1 was a four generation pedigree with 28 members, including 16 with congenital cataract (Figure 1A).FAMILY-2 was a three generation pedigree with 6 members,including 3 with congenital cataract (Figure 1B).
Figure 1 Pedigrees of two Chinese families with congenital cataract
Filled square: Male patient; Filled circle: Female patient;Empty symbol: Normal healthy individual; “/”: Deceased person.
Table 1 Clinical phenotypes and findings of patients in the congenital cataract families
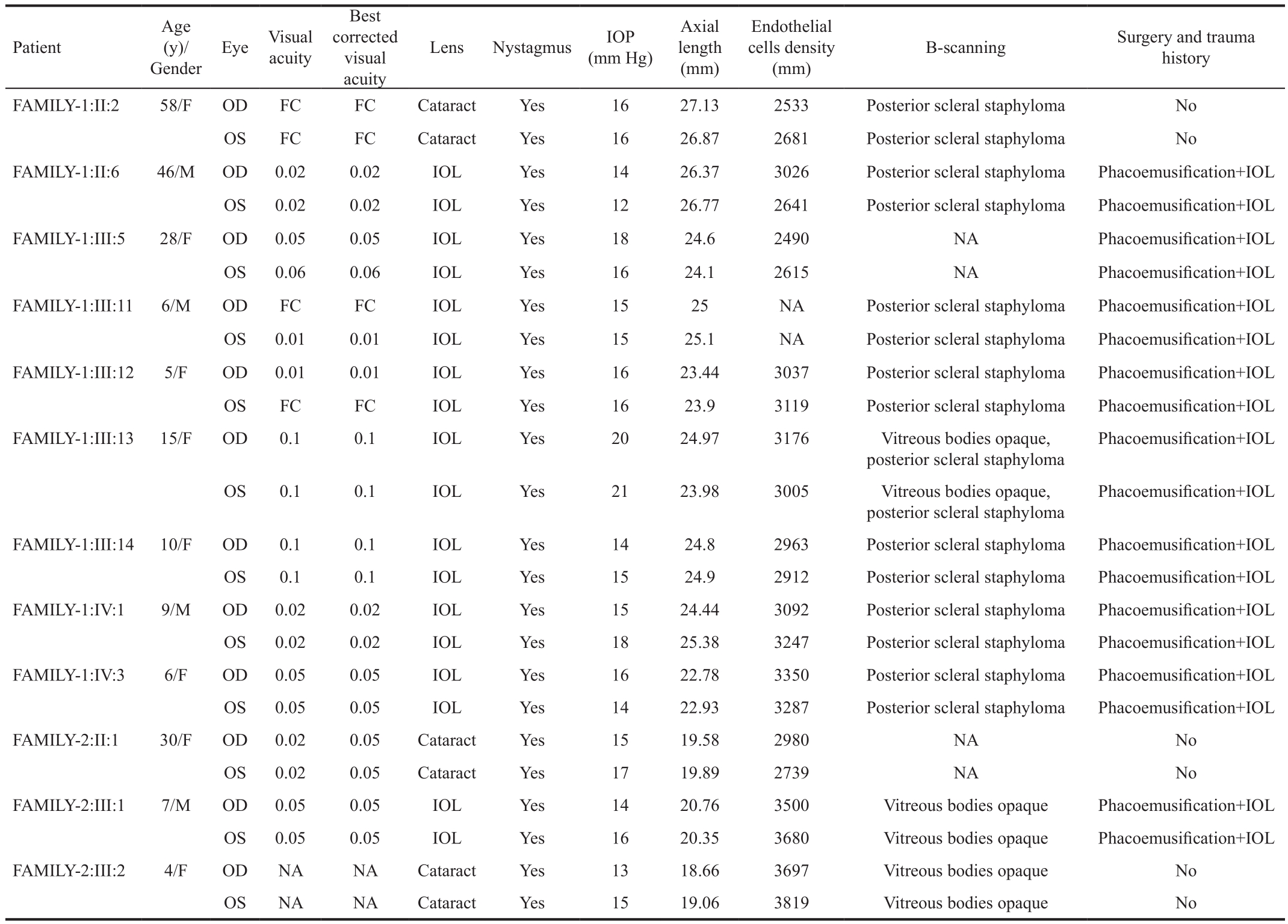
FC: Finger counting; F: Female; M: Male; IOL: Intraocular lens; NA: Not available; OD: Right eye; OS: Left eye; IOP: Intraocular pressure.
The patients in these two families had similar clinical phenotypes, such as congenital nuclear cataract and nystagmus.None of the two families had any other systemic diseases. The detailed clinical and ophthalmological findings of the patients of both families are shown in Table 1.
Identification ofCRYBB1/CRYBB2as a Candidate GeneNext, we aimed to identify pathogenic genes among 523 inheritable vision system-related genes by targeted exome sequencing (II:5 and II:1 in FAMILY-1 and 2, respectively).BWA was used for data analysis as proposed previously[15].The average sequencing depth of II:5 (FAMILY-1) was 58.34;72.21% of exon sequences were sequenced at least 10 times.The average sequencing depth of II:1 (FAMILY-2) was 60.89,and 84.33% of exon sequences were sequenced at least 10 times. NGS data are summarized in Table 2.
SNPs and indels were annotated, and filtered against 1000 Genome Project, the HapMap 8 database, the Single Nucleotide Polymorphism database, and the YH database. Since synonymous variants are unlikely to be pathogenic, we screened out nonsynonymous mutations (nonsense, missense and read-through),coding indels, and variants of splice donor and acceptor sites.The filtered data are shown in Table 3.
Table 2 Coverage parameters of targeted exome sequencing in the affected individuals, namely II:5 and II:1 in FAMILY-1 and 2,respectively
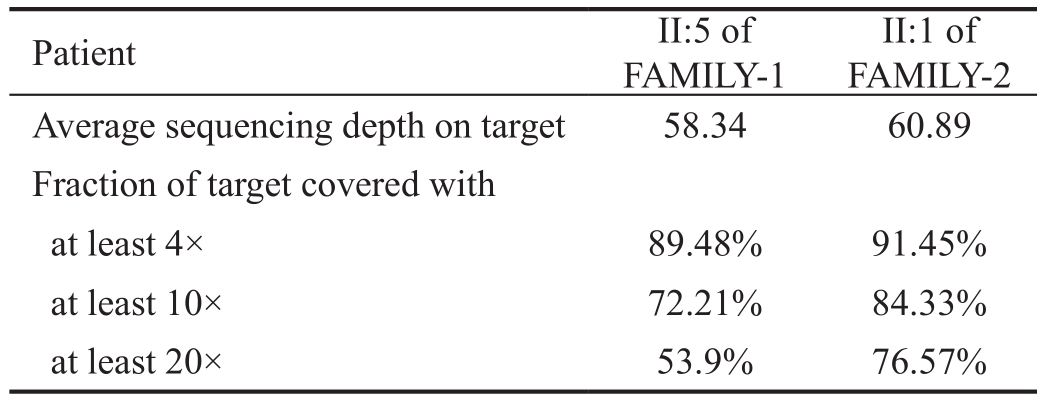
Table 3 SNPs and indels identified in the patients (II:5 and II:1 in FAMILY-1 and 2, respectively) by targeted NGS
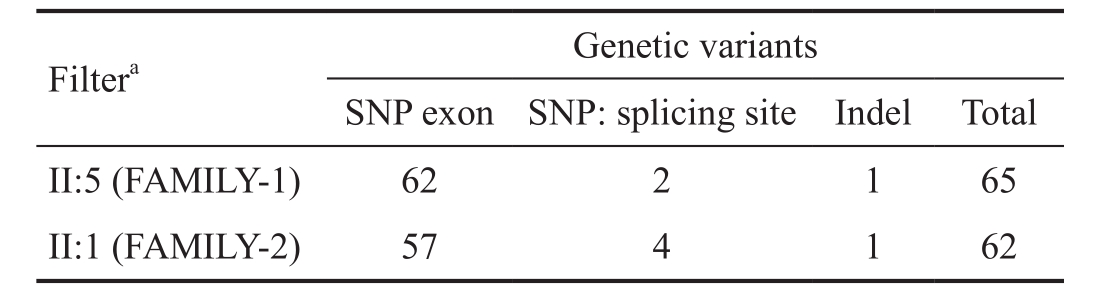
SNP: Single-nucleotide polymorphism;aNot in 1000 Genomes Project, the dbSNP, HapMap 8, or YH database.
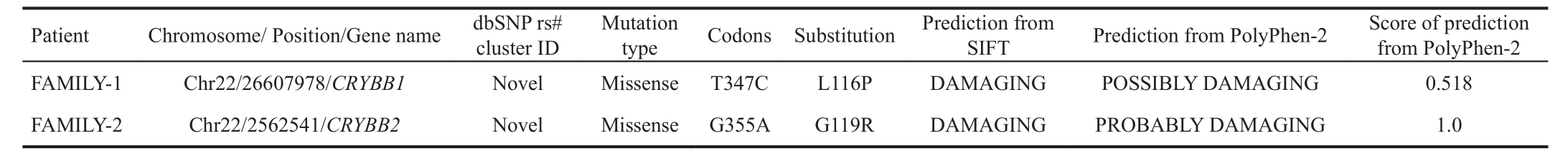
Sanger Sequencing to Verify the Candidate GeneCRYBB1/CRYBB2Sixty-two non-synonymous SNPs, 2 splicing sites,and 1 indel were selected by recommended filtering criteriain FAMILY-1. Only one SNP (Chr22:26607978) was cosegregated in FAMILY-1, in exon 4 ofCRYBB1; this was a T>C change (c.347T>C, p.L116P; Table 4, Figure 2A). This mutation was not found in 200 control individuals.
Table 4 SNPs and indels identified in the two congenital cataract families
SNP: Single-nucleotide polymorphism.
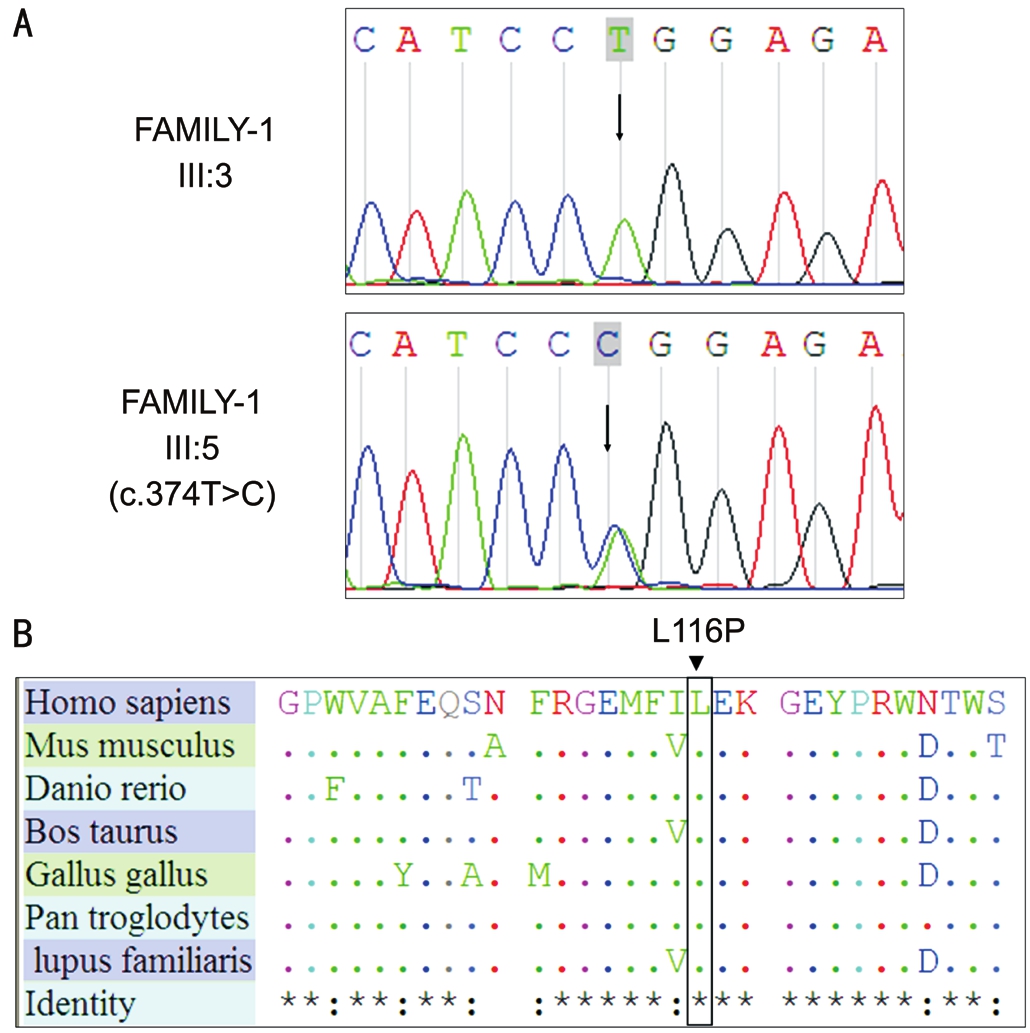
Figure 2 Chromatograms depicting theCRYBB1mutations of FAMILY-1
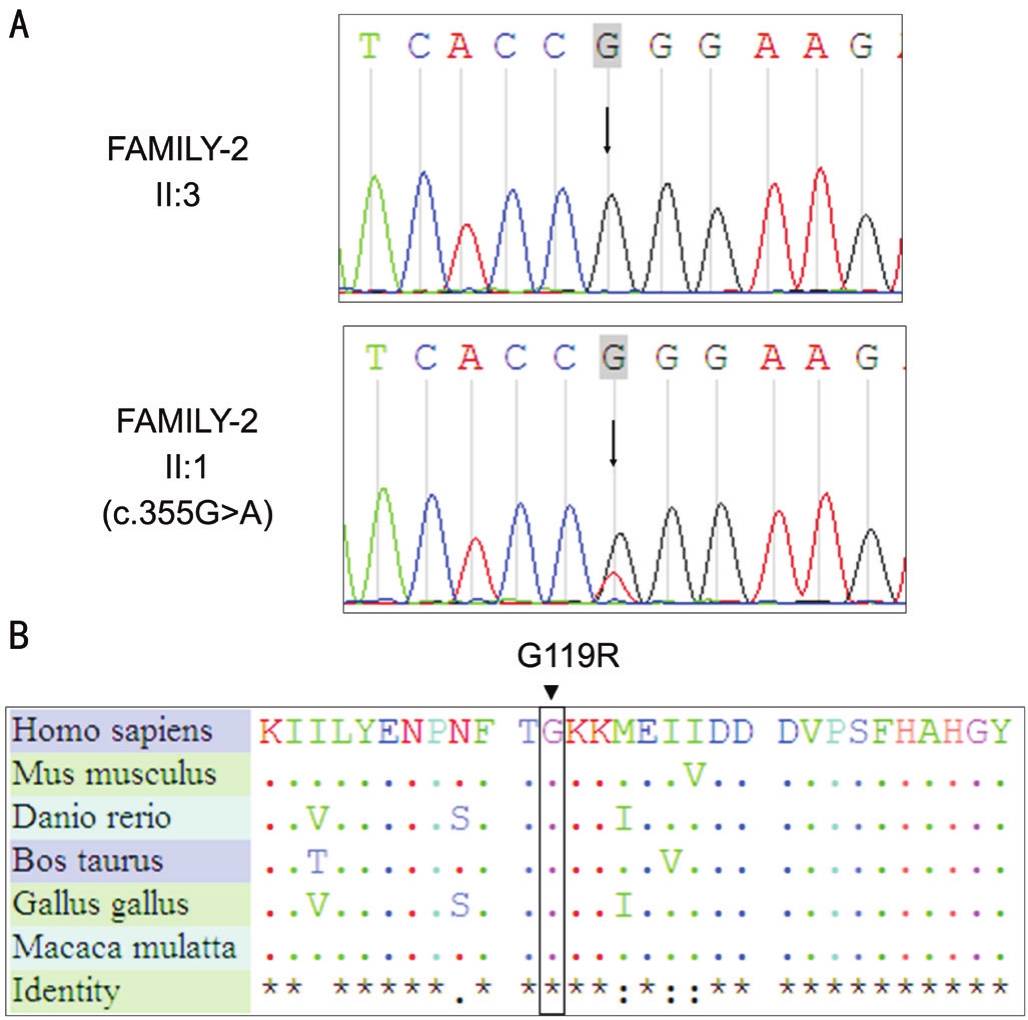
Figure 3 Chromatograms depicting theCRYBB2mutations of FAMILY-2
The p.L116P missense mutation was generated by c.347T>C.Homology analysis of leu116 loci of CRYBB1 proteins in different species indicated that these loci were highly conserved (Figure 2B). SIFT forecasted the p.L116P missense mutation to cause a deleterious change in CRYBB1function(Table 4).
Fifty-seven non-synonymous SNPs, 4 Splicing sites, and 1 indel were selected by recommended filtering criteria in FAMILY-2.Only one SNP (Chr22:2562541) was co-segregated in FAMILY-2, in exon 5 ofCRYBB2, a G>A change (c.355G>A,p.G119R; Table 4, Figure 3A). The mutation was not found in 200 control individuals.
The p.G119R missense mutation was generated by c.355G>A.Homology analysis of Gly119 loci ofCRYBB2proteins in different species indicated that these loci were highly conserved(Figure 3B). SIFT forcasted the p.G119R missense mutation to cause a deleterious change inCRYBB2function (Table 4).
The lens is a vascular organ whose transparency is important for maintaining normal function. Transparent lenses allow light to pass through and focus on the retina. The lens consists of two types of cells, including epithelial and fibrous cells. Lens fiber cells are differentiated from epithelial cells and contain high concentrations of soluble crystallin. The metabolic activity of mature fibroblasts is very limited, and most of the metabolism, biosynthesis, and active transport are accomplished by surface cells. Crystallins and a wide range of cell-cell communication systems are key factors in establishing and maintaining lens transparency. Abnormalities in lens cellsand/or crystallin may cause lens opacity, resulting in decreased vision and severe blindness[16].
In the vertebrate lens, β-crystallins are found as homomers or heteromers, which are thought to be important in maintaining the optical properties of the lens throughout an individual’s life[17-20].
The new heterozygous c.347T>C mutation found inCRYBB1(FAMILY-1), co-occurred with autosomal dominant cataract.CRYBB1represents the main β-crystallin subunit, and accounts for about a tenth of human non-membrane crystallins[21].The β-crystallin structure is considered to be critical in protein aggregation and orientation, with terminal arm loss altering their dimerization and causing cataract development[22]. Mackayet al[23]demonstrated that the G220X mutation inCRYBB1is related to autosomal dominant cataract, likely disrupting the 4thGreek key motif and destabilizingCRYBB1. Meanwhile, 3CRYBB1mutations related to autosomal dominance and/or autosomal recessive congenital cataracts were identified[24-26].
The newCRYBB2(FAMILY-2) mutation found in exon 5 (c.355G>A, p.G119R) co-occurred with autosomal dominant cataract. A total of 3 additionalCRYBB2mutations were described, namely Q155X, D128V, and W151C, also destabilizingCRYBB[27].
Reports describing gene mutations related to congenital cataracts often use whole exome sequencing with linkage assessment among relatives[28-33]; alternatively, a specific gene is screened in more individuals[34]. A total of 100 patients with congenital cataract were assessed by exon sequencing of fourteen genes,and 18 variant genes were detected, including 14 involving crystallins[35]. In another previous study of 61 genes in 74 nonrelated patients, 32 sporadic congenital cataract specific SNPs were obtained in 29 cases, including 31 with a hetero zygous pattern[36].
The NGS approach was employed in the present work for the assessment of 523 documented inheritable vision systemrelated genes in two families. Heterozygous mutations were found in pedigrees 1 (CRYBB1,exon 4; c.347T>C, p.L116P)and 2 (CRYBB2,exon 5; c.355G>A, p.G119R), exclusively cosegregated with diseased family members, and not found in 200 control Chinese individuals.
TheCRYBB1(c.347T>C) andCRYBB2(c.355G>A) mutations in this study are not found in previous reports. In comparison with whole exome sequencing, target region sequencing presents several advantages in assessing SNPs in sporadic cases due to low cost and easy analysis. It should be noted that the present sequencing strategy cannot identify unknown genes. Overall, these findings provide new insights regarding the etiological features of congenital cataract.
The abstract of this paper was published on theJournal of Annals of Anatomy. We would like to express our gratitude to the participating families. For the data set used to filter variants,we thank the Single Nucleotide Polymorphism database, 1000 Genome Project, HapMap 8 database, and YH database.
Conflicts of Interest:Chen P, None; Chen H, None; Pan XJ,None; Tang SZ, None; Xia YJ, None; Zhang H, None.
REFERENCES
1 Bermejo E, Martínez-Frías ML. Congenital eye malformations: clinicalepidemiological analysis of 1,124,654 consecutive births in Spain.Am J Med Genet1998;75(5):497-504.
2 Francis PJ, Berry V, Bhattacharya SS, Moore AT. The genetics of childhood cataract.J Med Genet2000;37(7):481-488.
3 Shiels A, Hejtmancik JF. Genetic origins of cataract.Arch Ophthalmol2007;125(2):165-173.
4 Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map.Mol Vis2010;16:2007-2015.
5 Shiels A, Hejtmancik JF. Genetics of human cataract.Clin Genet2013;84(2):120-127.
6 Hejtmancik JF. Congenital cataracts and their molecular genetics.Semin Cell Dev Biol2008;19(2):134-149.
7 Wang K, Wang B, Wang J, Zhou S, Yun B, Suo P, Cheng J, Ma X, Zhu S.A novel GJA8 mutation (p.I31T) causing autosomal dominant congenital cataract in a Chinese family.Mol Vis2009;15:2813-2820.
8 Mackay DS, Bennett TM, Culican SM, Shiels A. Exome sequencing identifies novel and recurrent mutations in GJA8 and CRYGD associated with inherited cataract.Hum Genomics2014;8:19.
9 Santana A, Waiswo M. The genetic and molecular basis of congenital cataract.Arq Bras Oftalmol2011;74(2):136-142.
10 Shiels A, Bennett TM, Knopf HL, Yamada K, Yoshiura K, Niikawa N, Shim S, Hanson PI. CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q.Am J Hum Genet2007;81(3):596-606.
11 Shiels A, Bennett TM, Knopf HL, Maraini G, Li A, Jiao X, Hejtmancik JF. The EPHA2 gene is associated with cataracts linked to chromosome 1p.Mol Vis2008;14:2042-2055.
12 Kaul H, Riazuddin SA, Shahid M, Kousar S, Butt NH, Zafar AU,Khan SN, Husnain T, Akram J, Hejtmancik JF, Riazuddin S. Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family.Mol Vis2010;16:511-517.
13 Ge XL, Zhang Y, Wu Y, Lv J, Zhang W, Jin ZB, Qu J, Gu F.Identification of a novel GJA8 (Cx50) point mutation causes human dominant congenital cataracts.Sci Rep2014;4:4121.
14 Li G, Ma L, Song C, Yang Z, Wang X, Huang H, Li Y, Li R, Zhang X,Yang H, Wang J, Wang J. The YH database: the first Asian diploid genome database.Nucleic Acids Res2009;37(Database issue):D1025-D1028.
15 Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform.Bioinformatics2009;25(14):1754-1760.
16 Beyer EC, Ebihara L, Berthoud VM. Connexin mutants and cataracts.Front Pharmacol2013;4:43.
17 Bloemendal H, de Jong W, Jaenicke R, Lubsen NH, Slingsby C,Tardieu A. Ageing and vision: structure, stability and function of lens crystallins.Prog Biophys Mol Biol2004;86(3):407-485.
18 Ajaz MS, Ma Z, Smith DL, Smith JB. Size of human lens betacrystallin aggregates are distinguished by N-terminal truncation of betaB1.J Biol Chem1997;272(17):11250-11255.
19 Slingsby C, Clout NJ. Structure of the crystallins.Eye (Lond)1999;13(Pt 3b):395-402.
20 Hejtmancik JF, Wingfield PT, Sergeev YV. Beta-crystallin association.Exp Eye Res2004;79:377-383.
21 Lampi KJ, Ma Z, Shih M, Shearer TR, Smith JB, Smith DL, David LL.Sequence analysis of betaA3, betaB3, and betaA4 crystallins completes the identification of the major proteins in young human lens.J Biol Chem1997;272(4):2268-2275.
22 Berbers GA, Boerman OC, Bloemendal H, de Jong WW. Primary gene products of bovine beta-crystallin and reassociation behavior of its aggregates.Eur J Biochem1982;128(2-3):495-502.
23 Mackay DS, Boskovska OB, Knopf HL, Lampi KJ, Shiels A. A nonsense mutation in CRYBB1 associated with autosomal dominant cataract linked to human chromosome 22q.Am J Hum Genet2002;71(5):1216-1221.
24 Willoughby CE, Shafiq A, Ferrini W, Chan LL, Billingsley G, Priston M, Mok C, Chandna A, Kaye S, Héon E. CRYBB1 mutation associated with congenital cataract and microcornea.Mol Vis2005;11:587-593.
25 Cohen D, Bar-Yosef U, Levy J, Gradstein L, Belfair N, Ofir R, Joshua S, Lifshitz T, Carmi R, Birk OS. Homozygous CRYBB1 deletion mutation underlies autosomal recessive congenital cataract.Invest Ophthalmol Vis Sci2007;48(5):2208-2213.
26 Wang J, Ma X, Gu F, Liu NP, Hao XL, Wang KJ, Wang NL, Zhu SQ. A missense mutation S228P in the CRYBB1 gene causes autosomal dominant congenital cataract.Chin Med J2007;120(9):820-824.
27 Berry V, Francis P, Kaushal S, Moore A, Bhattacharya S. Missense mutations in MIP underlie autosomal dominant ‘polymorphic’ and lamellar cataracts linked to 12q.Nat Genet2000;25(1):15-17.
28 Sun W, Xiao X, Li S, Guo X, Zhang Q. Exome sequencing of 18 Chinese families with congenital cataracts: a new sight of the NHS gene.PLoS One2014;9(6):e100455.
29 Narumi Y, Nishina S, Tokimitsu M, Aoki Y, Kosaki R, Wakui K,Azuma N, Murata T, Takada F, Fukushima Y, Kosho T. Identification of a novel missense mutation of MAF in a Japanese family with congenital cataract by whole exome sequencing: a clinical report and review of literature.Am J Med Genet A2014;164A(5):1272-1276.
30 González-Huerta LM, Messina-Baas O, Urueta H, Toral-López J,Cuevas-Covarrubias SA. A CRYGC gene mutation associated with autosomal dominant pulverulent cataract.Gene2013;529(1):181-185.
31 Yu Y, Yu Y, Chen P, Li J, Zhu Y, Zhai Y, Yao K. A novel MIP gene mutation associated with autosomal dominant congenital cataracts in a Chinese family.BMC Med Genet2014;15:6.
32 Wang H, Zhang T, Wu D, Zhang J. A novel beaded filament structural protein 1 (BFSP1) gene mutation associated with autosomal dominant congenital cataract in a Chinese family.Mol Vis2013;19:2590-2595.
33 Reis LM, Tyler RC, Muheisen S, Raggio V, Salviati L, Han DP,Costakos D, Yonath H, Hall S, Power P, Semina EV. Whole exome sequencing in dominant cataract identifies a new causative factor,CRYBA2, and a variety of novel alleles in known genes.Hum Genet2013;132(7):761-770.
34 Dave A, Laurie K, Staffieri SE, Taranath D, Mackey DA, Mitchell P,Wang JJ, Craig JE, Burdon KP, Sharma S. Mutations in the EPHA2 gene are a major contributor to inherited cataracts in South-Eastern Australia.PLoS One2013;8(8):e72518.
35 Kumar M, Agarwal T, Kaur P, Kumar M, Khokhar S, Dada R.Molecular and structural analysis of genetic variations in congenital cataract.Mol Vis2013;19:2436-2450.
36 Li D, Wang S, Ye H, Tang Y, Qiu X, Fan Q, Rong X, Liu X, Chen Y, Yang J, Lu Y. Distribution of gene mutations in sporadic congenital cataract in a Han Chinese population.Mol Vis2016;22:589-598.
Citation:Chen P, Chen H, Pan XJ, Tang SZ, Xia YJ, Zhang H.Novel mutations inCRYBB1/CRYBB2identified by targeted exome sequencing in Chinese families with congenital cataract.Int J Ophthalmol2018;11(10):1577-1582
DOl:10.18240/ijo.2018.10.01
Accepted:2018-05-08
Received:2017-11-30
Correspondence to:Yu-Jun Xia. Qingdao University, 308 Ningxia Road, Qingdao 266071, Shandong Province, China.xiayujun62@163.com; Hui Zhang. Jinan Second People’s Hospital, 148 Jingyi Road, Jinan 250022, Shandong Province,China. zhanglinsay@126.com.
Co-first authors:Peng Chen and Hao Chen
Foundations:Supported by China Postdoctoral Science Foundation Funded Project (No.2017M612211); Shandong Provincial Natural Science Foundation (No.ZR2018MH016);Qingdao Postdoctoral Application Research Project(No.40518060071); Medical Program of Shandong Province(No.2016WS0265); Qingdao Science and Technology Plan (No.16-6-2-14-nsh); and Shandong Province Higher Educational Science and Technology Program (No.J17KA235).