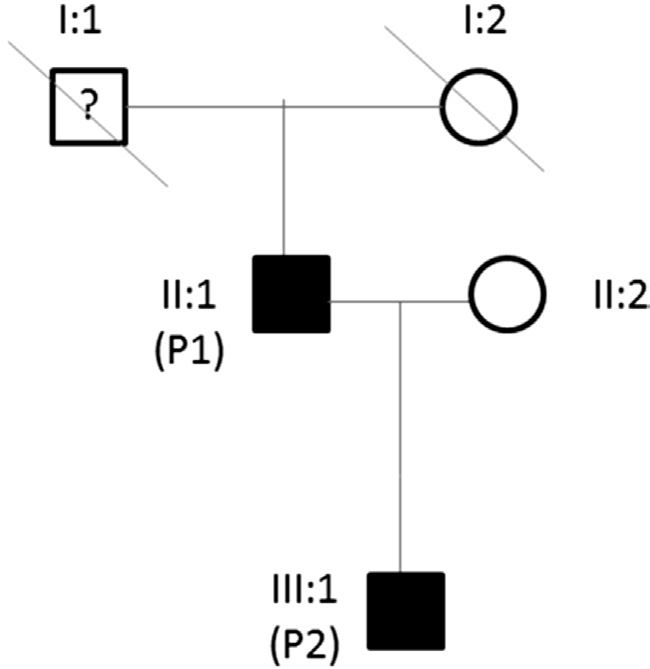
Figure 1 Family pedigree.
Cone dystrophies (CD) and cone-rod dystrophies (CRD)are a group of genetic disorders, which demonstrate a large degree of heterogeneity and severity. The most frequent mode of inheritance is autosomal dominant, although autosomal recessive and X-linked recessive modes of inheritance have also been reported[1]. The main symptoms are decreased central visual acuity (VA), markedly decreased color vision, hemeralopia, nystagmus and loss of peripheral vision[2].In pure cone dystrophies, only cone function is affected, while rod function remains intact. The photopic electroretinograms(ERG) demonstrates abnormalities but the scotopic ERG is grossly normal. Conversely, in cone-rod dystrophies, patients demonstrate features suggestive of rod dysfunction as well. In CRD, both photopic and scotopic ERGs will be abnormal[1-3]. It is rare to have a pure cone dystrophy because of the reciprocal relationship between the cone and rod system[3].
The usual natural history of CRD starts initially by forming some non-speci fic retinal pigment epithelial (RPE) granularity and mottling at the macular level. As the disease progresses,a typical bull’s eye lesion develops, but not universally. Endstage disease with photoreceptor degeneration and RPE loss will result in geographic atrophy[3]. There is a wide range of genes implicated in the pathogenesis of CRD. The most common ones areSEMA4A,AIPL1, CRX, GUCA1A,GUCY2D, PITPNM3, PRPH2, PROM1, RIMS1andUNC119[4].
Both family members were found to have mutation in the retinal guanylyl cyclase 1 gene (also known as guanylate cyclase 2D/GUCY2D), which is known to be implicated in the autosomal dominant form of cone/cone-rod dystrophy. Retinal guanylyl cyclase 1 is an enzyme expressed within the retina responsible for the conversion of guanosine 5’-triphosphate to cyclic guanosine monophosphate (cGMP)[4]. Like other membrane guanylyl cyclases, this enzyme has a hydrophobic amino-terminal signal sequence followed by a large extracellular domain, a single membrane spanning domain,a kinase homology domain, and a guanylyl cyclase catalytic domain. In contrast to other membrane guanylyl cyclases,this enzyme is not activated by natriuretic peptides. Retinal guanylyl cyclase 1 helps photoreceptors return to their darkadapted state after light exposure; cGMP plays a significant role as the second messenger molecule in the phototransduction cascade by keeping the voltage-gated sodium and calcium channels of photoreceptors open. Photoactivation leads to conversion of cGMP to guanosine 5’-monophophate by phosphodiesterase and this results in the closure of voltagegated sodium and calcium channels and to hyperpolarization of the photoreceptor outer segments. When the concentration of calcium cations is reduced, retinal guanylyl cyclase 1 restores the levels of cGMP and this allows the reopening of the relevant channels. Restoration of cGMP levels is achieved by the presence of the guanylate cyclase-activating protein[4].Mutations inGUCY2Dgene have been described in cone-rod dystrophy-6 and Leber congenital amaurosis[5].
In this manuscript, we describe the long-term clinical and multimodal imaging findings over the course of 13y in two family members diagnosed withGUCY2Dcone dystrophy.To the best of our knowledge, this is the longest follow-up described so far in literature.
All procedures were compliant and consistent with the tenets of the Declaration of Helsinki. Informed consent was obtained from all individual participants included in this retrospective study. A retrospective review of the electronic records of two family members (father and son) was conducted at the Eye Unit of University Hospital Southampton National Healthcare System Foundation Trust, UK. Both patients were followedup annually at the Eye Unit for the last 13y and had a full past medical, ophthalmic and genetic history taken during the initial presentation. Annual follow-up visits were conducted including multimodal imaging. Optical coherence tomography(OCT) scans were obtained with the use of Triton/OCT-2000(Topcon Ltd, Tokyo, Japan), whereas fundus auto fluorescence(FAF) images were taken using Spectralis (Heidelberg Engineering, Heidelberg, Germany). Goldmann visual field(GVF) testing was utilised to monitor the progression of central field loss. Flash and pattern ERGs were recorded using corneal Dawson-Trick-Litzkow (DTL) thread electrodes.Flash ERGs were recorded after dilatation in compliance with International Society for Clinical Electrophysiology of Vision standards[6]. Occipital full-field checkerboard reversal visual evoked potentials (VEPs) were recorded to stimulus check sizes ranging from 10 to 120min of arc.
Figure 1 Family pedigree.
Both patients underwent genetic testing: the participants underwent whole exome sequencing in order to identify the genetic cause of their CD. DNA was isolated from blood,exome enrichment performed using the Agilent SureSelect Human All Exon V5 kit (© Agilent Technologies, Inc), and sequencing performed on the Illumina HiSeq 2000 platform(© Illumina Inc®). Data analysis was performed as previously described[7]. Genetics variants were filtered to identify variants present in both individuals within candidate genes identified through the Human Gene Mutation Database (namelyABCA4,CACNA2D4, CNGA3, CNGB3, CRB1, CRX, GUCA1A,GUCY2D, KCNV2, MERTK, orf15, PDE6C, PDE6H,PITPNM3 & PRPH2).
Genetic TestingExome sequencing identified the heterozygous variantGUCY2D:c.2512C>T:p.Arg838Cys(rs61750172, also known as R838C) in both patients (Figure 1).This mutation has been previously reported to cause cone-rod dystrophy 6 (CORD6; OMIM #601777)[8]. For abbreviation purposes, the father has been allocated the symbol P1 in generation II, whereas the son has been allocated the symbol P2 in generation III. There was also a history of eye problems in patient I1. There was insuf ficient data in past medical history to con firm a formal diagnosis of CD; hence the question mark symbol (Figure 1).
Clinical FindingsThe cumulative clinical features for each patient are summarized in Table 1. Both patients exhibited decline in VA combined with hemeralopia in adolescence but neither of them complained of nyctalopia. In addition, there were no significant media opacities to account for decline in VA in both of our patients. Figure 2 demonstrates the fluctuation of best-corrected visual acuity (BCVA) in both family members over the 13y follow-up at Southampton Eye Unit.
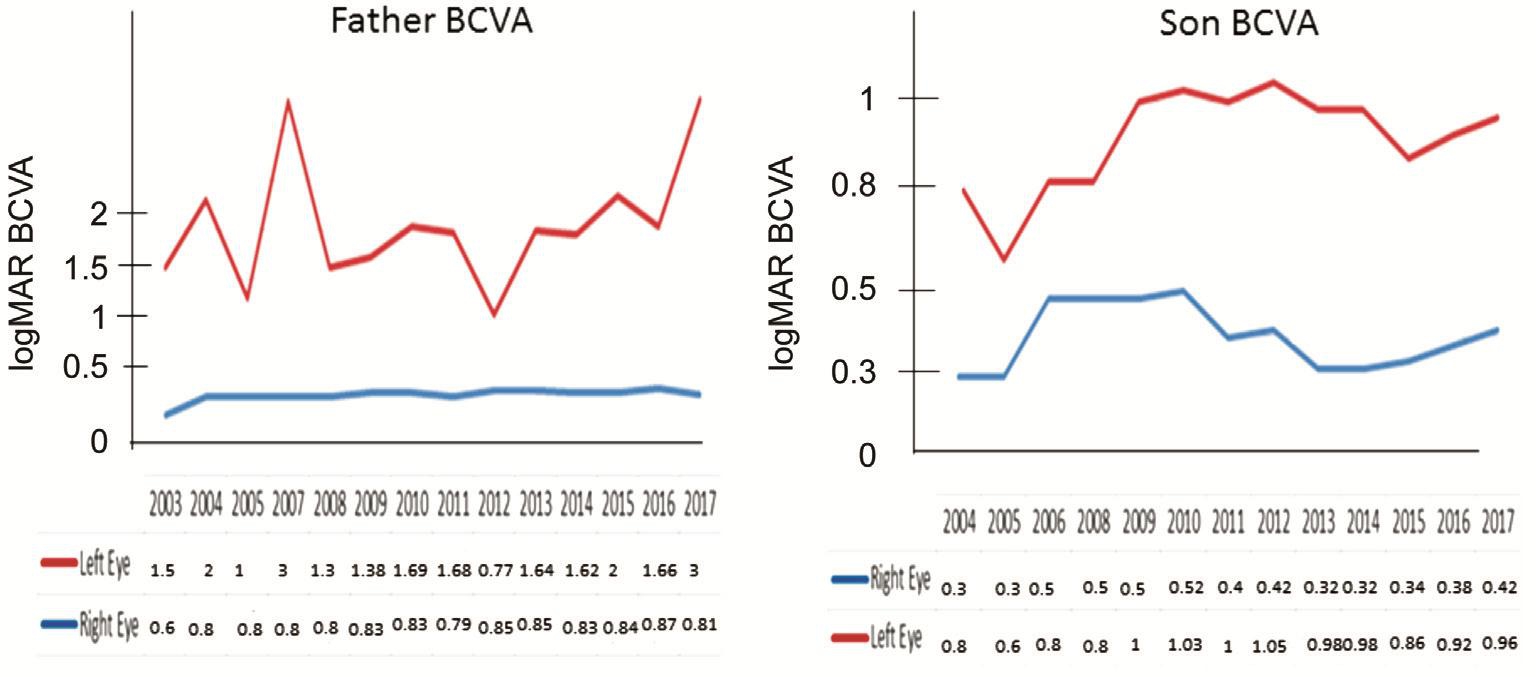
Figure 2 Changes in BCVA for both patients from initial presentation in 2004 until 2017.
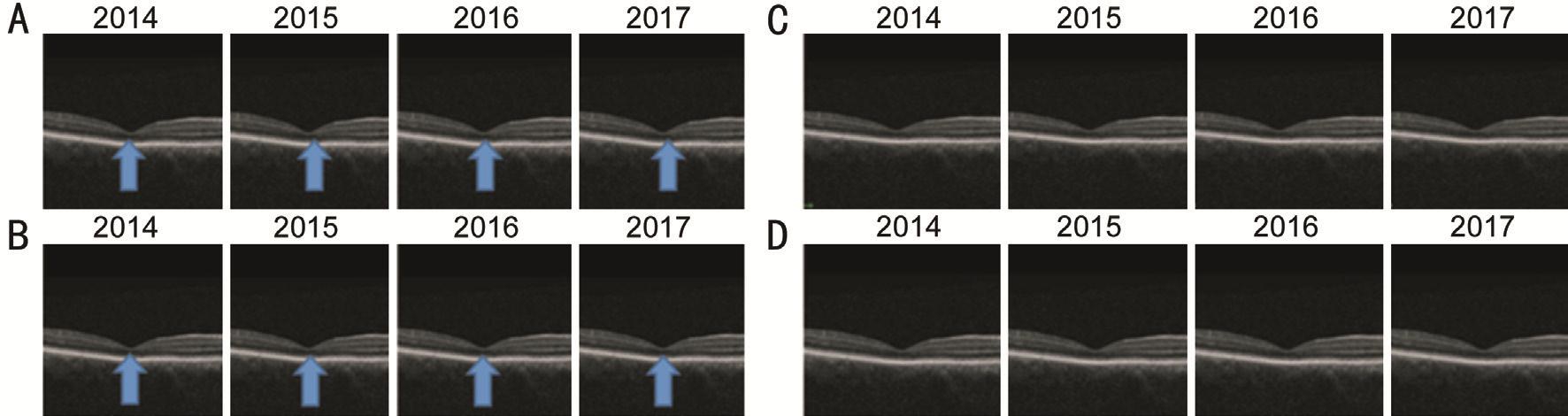
Figure 3 Serial OCT images both patients A: P1’s right eye; B: P1’left eye; C: P2’s right eye; D: P2’s left eye. Progressive atrophy of the ellipsoid layer but no breaks in the continuity of the ellipsoid layers on the OCT images.
Table 1 Cumulative table summarizing the clinical features in both family members
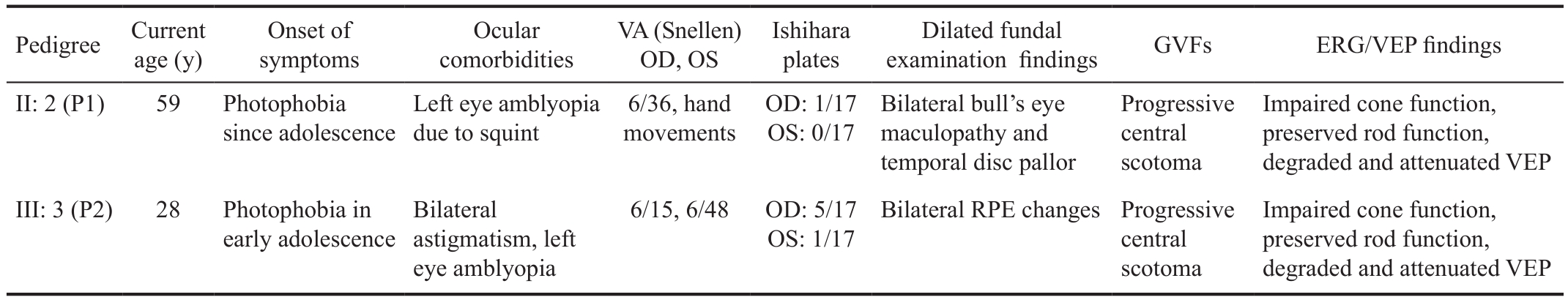
VA: Visual acuity; GVFs: Goldmann visual fields; ERG: Electroretinograms; VEP: Visual evoked potentials. Neither of the affected family members reported symptoms of nyctalopia or had signi ficant cataracts.
Pedigree Current age (y)Onset of symptoms Ocular comorbidities VA (Snellen)OD, OS Ishihara plates Dilated fundal examination findings GVFs ERG/VEP findings II: 2 (P1) 59 Photophobia since adolescence Impaired cone function,preserved rod function,degraded and attenuated VEP Impaired cone function,preserved rod function,degraded and attenuated VEP III: 3 (P2) 28 Photophobia in early adolescence Left eye amblyopia due to squint 6/36, hand movements OD: 1/17 OS: 0/17 Bilateral bull’s eye maculopathy and temporal disc pallor Progressive central scotoma Bilateral astigmatism, left eye amblyopia 6/15, 6/48 OD: 5/17 OS: 1/17 Bilateral RPE changesProgressive central scotoma
Multimodal ImagingThe macular OCT scans of P1 showed progressive loss of the ellipsoid layer at the level of the fovea with gradual thinning and atrophy of the adjacent retinal tissue and reverse shadowing due to cone and RPE cell loss(Figure 3A, 3B). FAF showed a central annular area of hypoautofluorescence corresponding to macular atrophy and RPE loss with a surrounding ring of hyper-autofluorescence(hyper-AF) indicating the transition zone between normal and abnormal retina (Figure 4A, 4B). These changes have occurred in both eyes but the left eye appears to be more affected than the right. The macular OCT scan of the right eye of P2 demonstrated gradual macular thinning and atrophy, whereas the macular structure in the left eye remained relatively stable.Unlike the father’s OCT scans, there was no disruption of the ellipsoid layer (Figure 3C, 3D). FAF images of the son showed features suggestive of bilateral foveolar hyper-AF. The hyper-AF involving the central foveolar area which can be seen in Figure 4C and 4D, are similar to the changes previously reported in type-2 idiopathic macular telangiectasia[9]. FAF findings also demonstrated RPE granular changes.
ElectrophysiologyThe father’s flash ERGs showed wellpreserved rod function (amplitude of responses smaller than average but within normal range) but significantly impaired cone function. Pattern ERGs as well occipital pattern VEPs were attenuated and degraded indicating reduced macular function. The son’s cone responses were of borderline normal amplitude on initial presentation but became significantly degraded ten months later suggesting cone dysfunction(Figure 5). Rod responses were normal. Pattern ERGs and occipital pattern VEPs were signi ficantly degraded indicating reduced macular function.

Figure 4 Auto fluorescence images from both patients A, B: Father (P1): central area of hypo-auto fluorescence, left worse than right; C,D: Son (P2): bilateral foveolar hyper-AF. The hyper-AF changes may be a consequence of decreased foveal pigment density and secondarily reduced masking effect of the RPE fluorescence.
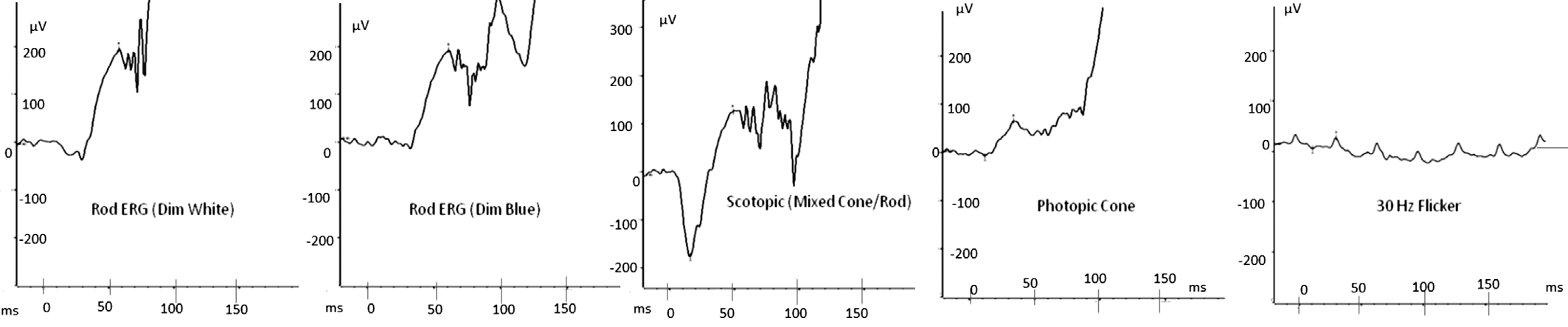
Figure 5 P2’s ERG responses It shows P2’s repeated ERG responses a few months after initial presentation. There was a signi ficant reduction in the amplitude of the cone mediated responses, which were more degraded compared to the initial ERGs.
So far, 223 mutations in theGUCY2Dgene have been described. TheArg838Cys (R838C)mutation described in both of our patients has been previously reported by Kellsellet al[10]in 1998 in cone dystrophy 6. It is reported to cause a milder clinical phenotype compared to other mutations in theGUCY2Dgene[11]. The mild phenotype of this particular mutation has been described by others in the past[12-13].OtherGUCY2Dmutations on the same codon (R838S,R838H, R838P, R838G) can lead to a more aggressive clinical picture[14-15]. Based on ERG recordings, two major types of cone-rod dystrophy were differentiated according to the phenotypic classification by Szlyket al[16]. In type 1,cone amplitudes were reduced to a greater degree than rod amplitudes, while in type 2, cone and rod ERG amplitudes were reduced in equal proportion. According to the phenotypic classification by Szlyket al[16], both of our patients could be classi fied as phenotype 1a.
The father presented to the Ophthalmology Department with bilateral bull’s eye maculopathy and mild temporal disc pallor. Bull’s eye maculopathy can be caused by genetically inherited conditions or toxic retinopathies, hence it is not disease speci fic for cone/cone-rod dystrophy[3]. Disc pallor is also a non-specific finding but has been reported previously in a patient with cone dystrophy[17], who had normal to near normal VA and color vision and abnormal peripheral cone function. However, in the father’s case, the macula function was already compromised and the peripheral retina was normal. His son exhibited non-speci fic RPE granular changes but no other signi ficant abnormalities. Bull’s eye maculopathy was not observed in the son’s case con firming that bull’s eye maculopathy is not a universal sign[3]. This may be merely due to the chronicity of the disorder in his father. Moreover, the presence of a bull’s eye maculopathy does not always correlate accurately with the extent of retinal dysfunction[18]. All the above observations confirm that the diagnosis of CD/CRD cannot rely exclusively on fundoscopy due to the non-speci fic clinical findings[19].
On spectral-domain optical coherence tomography (SDOCT), the father exhibited progressive loss of the ellipsoid layer and gradual thinning and atrophy of the parafoveal retinal tissue and reverse shadowing due to cone and RPE cell loss. There was also obscurity at the level of the external limiting membrane (EML). This is consistent with the findings of others[19-23]. His son, however, had thinning but no loss of the ellipsoid layer. FAF imaging from the father’s fundus was consistent with the OCT findings. Furthermore,the surrounding hyper-AF around the annular area of hypoautofluorescence suggests gradual deposition of lipofuscin material, a byproduct of the photoreceptor cell visual cycle and RPE metabolism. Lipofuscin accumulation can be toxic to the RPE and photoreceptor cells and this can lead to death of RPE and photoreceptors and that can cause further thinning and atrophy of the macula[24]. The FAF findings from the father’s fundus are consistent with observations of another paper[25].The RPE granular changes observed in the son’s fundus were also observed by FAF. Hence, FAF is useful as an adjuvant means of imaging when SD-OCT cannot detect subtle RPE or retinal abnormalities. FAF images showing a subtle bilateral hyper-AF signal mimicking changes that were previously described in Type 2 Macular Telangiectasia might be a reliable early indicator of the disease especially when ERGs are found to be border-line normal as in P2 in this case series[7].
Only one paper by Choet al[26]has attempted to describe in depth and classify the different types of structural retinal abnormalities in patients with CD/CRD. This was a five year observational follow-up in 15 patients with cone dystrophy.Prior to this study, Hoodet al[21]reported decreased intensity in the ellipsoid layer in 6 patients with cone dystrophy.Birchet al[27]reported that the thickness of the outer nuclear layer and the sum of thickness of the RPE and outer segment correlated well with visual field sensitivity[20,24]. However,neither paper described the structural changes of the retina in patients with cone dystrophies.
Choet al[26]divided the morphological changes in the retinal structure in cone dystrophy patients into four different categories: 0, 1, 2 and 3. Category 0 exhibited no structural abnormalities, whereas category 1 showed foveal ellipsoid layer loss and obscurity of the border between the ellipsoid band and ELM. Category 2 showed foveal thinning and focal foveal ellipsoid layer disruption with an intact ELM. Finally,category 3 showed foveal thickening and perifoveal disruption of the ellipsoid layer.
Based on this classi fication, the father demonstrated changes matching category 1. The son did have foveal thinning but no disruption of the ellipsoid layer, hence he could potentially be classi fied as category 0. In the Choet al’s[26]paper, it was observed that category 0 patients were younger than the other categories, although this observation was not proven to be statistically significant. Ageing is likely to be a contributing factor to disease progression with subsequent disruption of the photoreceptor outer segment/ellipsoid layer.
Moreover, in the paper by Choet al[26], only one patient was found to meet the category 3 criteria. The authors formed the hypothesis that the thickening of the fovea could be attributed to the gradual deposition of tissue remnants of the unhealthy and gradually dying photoreceptors. This SD-OCT finding has been described previously in patients withperipherin/RDSgene mutations[28]. However, in patients with CD due toGUCY2Dmutations, category 1 abnormalities have been previously described (GUCY2D Arg838His). The difference to the case described by Kimet al[29](GUCY2D Arg838His)is that our patients had theGUCY2DArg838Cysmutation.Nevertheless, the morphological features on SD-OCT are similar.
Electrophysiological testing is arguably the most diagnostic test, should CD/CRD be suspected. Fundus examination can show non-speci fic changes and the multimodal imaging findings in patients with cone dystrophy are quite heterogeneous and therefore fundoscopy, SD-OCT and FAF are not diagnostic.This is also supported by Choet al[26], who observed that category 0 patients had a significantly affected ERG, while no structural abnormalities were observed. In addition, it is obvious that electrophysiological responses do not correlate well with SD-OCT findings. Thus, ERG and VEP can con firm the macula/cone dysfunction much earlier than other imaging modalities and therefore they both are an irreplaceable adjuvant diagnostic tool for any Medical Retina Specialist in the diagnosis and management of patients diagnosed with CD/CRD.
To the best of our knowledge, this is the longest duration of follow-up of patients with CD associated with mutations inGUCY2D. We describe the progression of the disease based on VA and multimodal imaging. Electrophysiological testing is most useful for the clinical diagnosis of CD/CRD, while SDOCT and FAF imaging are both useful for monitoring disease progression and genotype-phenotype correlations can be identi fied by molecular analysis.
Authors would like to express gratitude to the patients for their involvement in this study. We thank Dr. David Bunyan,Senior Clinical Scientist, SalisburyNational Healthcare System Foundation Trust for con firmatory genetic testing and Ms. Angela Cree, Senior Research Manager, University of Southampton for overall project coordination. We thank Fight against Blindness Charity Organization for supporting this project.
Foundation:Supported by Fight Against Blindness Charity Organization.
Conflicts of Interest: Tsokolas G,None;Almuhtaseb H,None;Griffiths H,None;Shawkat F,None;Pengelly RJ,None;Ennis S,None;Lotery A,None.
1 Aboshiha J, Dubis AM, Carroll J, Hardcastle AJ, Michaelides M. The cone dysfunction syndromes.Br J Ophthalmol2016;100(1):115-121.
2 Thiadens AA, Phan TM, Zekveld-Vroon RC,et al. Clinical course,genetic etiology, and visual outcome in cone and cone-rod dystrophy.Ophthalmology2012;119(4):819-826.
3 Kanski J.Clinical ophthalmology, a systematic approach. Elsevier 2015.
4 Nong E, Lee W, Merriam JE, Allikmets R, Tsang SH. Disease progression in autosomal dominant cone-rod dystrophy caused by a novel mutation (D100G) in the GUCA1A gene.Doc Ophthalmol2014;128(1):59-67.
5 Boye SE. Leber congenital amaurosis caused by mutations in GUCY2D.Cold Spring Harb Perspect Med2014;5(1):a017350.
6 McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, Bach M. ISCEV standard for full-field clinical electroretinography (2015 update).Doc Ophthalmol2015;130(1):1-12.
7 Pengelly RJ, Upstill-Goddard R, Arias L, Martinez J, Gibson J, Knut M,Collins AL, Ennis S, Collins A, Briceno I. Resolving clinical diagnoses for syndromic cleft lip and/or palate phenotypes using whole-exome sequencing.Clin Genet2015;88(5):441-449.
8 Jiang F, Xu K, Zhang X, Xie Y, Bai F, Li Y. GUCY2D mutations in a Chinese cohort with autosomal dominant cone or cone-rod dystrophies.Doc Ophthalmol2015;131(2):105-114.
9 Toto L, Di Antonio L, Mastropasqua R, Mattei PA, Carpineto P, Borrelli E, Rispoli M, Lumbroso B, Mastropasqua L. Multimodal imaging of macular telangiectasia type 2: focus on vascular changes using optical coherence tomography angiography.Invest Ophthalmol Vis Sci2016;57(9):OCT268-276.
10 Kelsell RE, Gregory-Evans K, Payne AM, Perrault I, Kaplan J, Yang RB, Garbers DL, Bird AC, Moore AT, Hunt DM. Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy.Hum Mol Genet1998;7(7):1179-1184.
11 Wilkie SE, Newbold RJ, Deery E, Walker CE, Stinton I, Ramamurthy J, Hurley JB, Bhattacharya SS, Warren MJ, Hunt DM. Functional characterization of missense mutations at codon 838 of retinal guanylate cyclase correlates with disease severity in patients with autosomal dominant cone-rod dystrophy.Hum Mol Genet2000;9(20):3065-3073.
12 Downes SM, Payne AM, Kelsell RE, Fitzke FW, Holder GE, Hunt DM, Moore AT, Bird AC. Autosomal dominant cone-rod dystrophy with mutations in the guanylate cyclase 2D gene encoding retinal guanylate cyclase-1.Arch Ophthalmol2001;119(11):1667-1673.
13 Van Ghelue M, Eriksen HL, Ponjavic V, Fagerheim T, Andreasson S,Forsman-Semb K, Sandgren O, Holmgren G, Tranebjaerg L. Autosomal dominant cone-rod dystrophy due to a missense mutation (R838C) in the guanylate cyclase 2D gene (GUCY2D) with preserved rod function in one branch of the family.Ophthalmic Genet2000;21(4):197-209.
14 Gregory-Evans K, Kelsell RE, Gregory-Evans CY, Downes SM,Fitzke FW, Holder GE, Simunovic M, Mollon JD, Taylor R, Hunt DM,Bird AC, Moore AT. Autosomal dominant cone-rod retinal dystrophy(CORD6) from heterozygous mutation of GUCY2D, which encodes retinal guanylate cyclase.Ophthalmology2000;107(1):55-61.
15 Mukherjee R, Robson AG, Holder GE, Stockman A, Egan CA, Moore AT, Webster AR. A detailed phenotypic description of autosomal dominant cone dystrophy due to a de novo mutation in the GUCY2D gene.Eye(Lond)2014;28(4):481-487.
16 Szlyk JP, Fishman GA, Alexander KR, Peachey NS, Derlacki DJ.Clinical subtypes of cone-rod dystrophy.Arch Ophthalmol1993;111(6):781-788.
17 Ito N, Kameya S, Gocho K, Hayashi T, Kikuchi S, Katagiri S, Gekka T, Yamaki K, Takahashi H, Tsuneoka H. Multimodal imaging of a case of peripheral cone dystrophy.Doc Ophthalmol2015;130(3):241-251.
18 Kurz-Levin MM, Halfyard AS, Bunce C, Bird AC, Holder GE. Clinical variations in assessment of bull’s-eye maculopathy.Arch Ophthalmol2002;120(5):567-575.
19 Kellner U, Kellner S. Clinical findings and diagnostics of cone dystrophy.Ophthalmologe2009;106(2):99-108.
20 Mallipatna A, Vinekar A, Jayadev C, Dabir S, Sivakumar M, Krishnan N, Mehta P, Berendschot T, Yadav NK. The use of handheld spectral domain optical coherence tomography in pediatric ophthalmology practice: our experience of 975 infants and children.Indian J Ophthalmol2015;63(7):586-593.
21 Hood DC, Zhang X, Ramachandran R, Talamini CL, Raza A,Greenberg JP, Sherman J, Tsang SH, Birch DG. The inner segment/outer segment border seen on optical coherence tomography is less intense in patients with diminished cone function.Invest Ophthalmol Vis Sci2011;52(13):9703-9709.
22 Thomas MG, Kumar A, Kohl S, Proudlock FA, Gottlob I. Highresolution in vivo imaging in achromatopsia.Ophthalmology2011;118(5):882-887.
23 Leng T, Marmor MF, Kellner U, Thompson DA, Renner AB, Moore W,Sowden JC. Foveal cavitation as an optical coherence tomography finding in central cone dysfunction.Retina2012;32(7):1411-1419.
24 Yung M, Klufas MA, Sarraf D. Clinical applications of fundus auto fluorescence in retinal disease.Int JRetina Vitreous2016;2:12.
25 Wang NK, Chou CL, Lima LH, Cella W, Tosi J, Yannuzzi LA, Tsang SH. Fundus autofluorescence in cone dystrophy. Doc Ophthalmol2009;119(2):141-144.
26 Cho SC, Woo SJ, Park KH, Hwang JM. Morphologic characteristics of the outer retina in cone dystrophy on spectral-domain optical coherence tomography.Korean J Ophthalmol2013;27(1):19-27.
27 Birch DG, Wen Y, Locke K, Hood DC. Rod sensitivity, cone sensitivity, and photoreceptor layer thickness in retinal degenerative diseases.Invest Ophthalmol Vis Sci2011;52(10):7141-7147.
28 Duncan JL, Talcott KE, Ratnam K, Sundquist SM, Lucero AS, Day S, Zhang Y, Roorda A. Cone struc ture in retinal degeneration associated with mutations in the peripherin/RDS gene.Invest Ophthalmol Vis Sci2011;52(3):1557-1566.
29 Kim BJ, Ibrahim MA, Goldberg MF. Use of spectral domain OCT to visualize photoreceptor abnormalities in cone/rod dystrophy-6.Retin Cases Brief Rep2011;5(1):56-61.