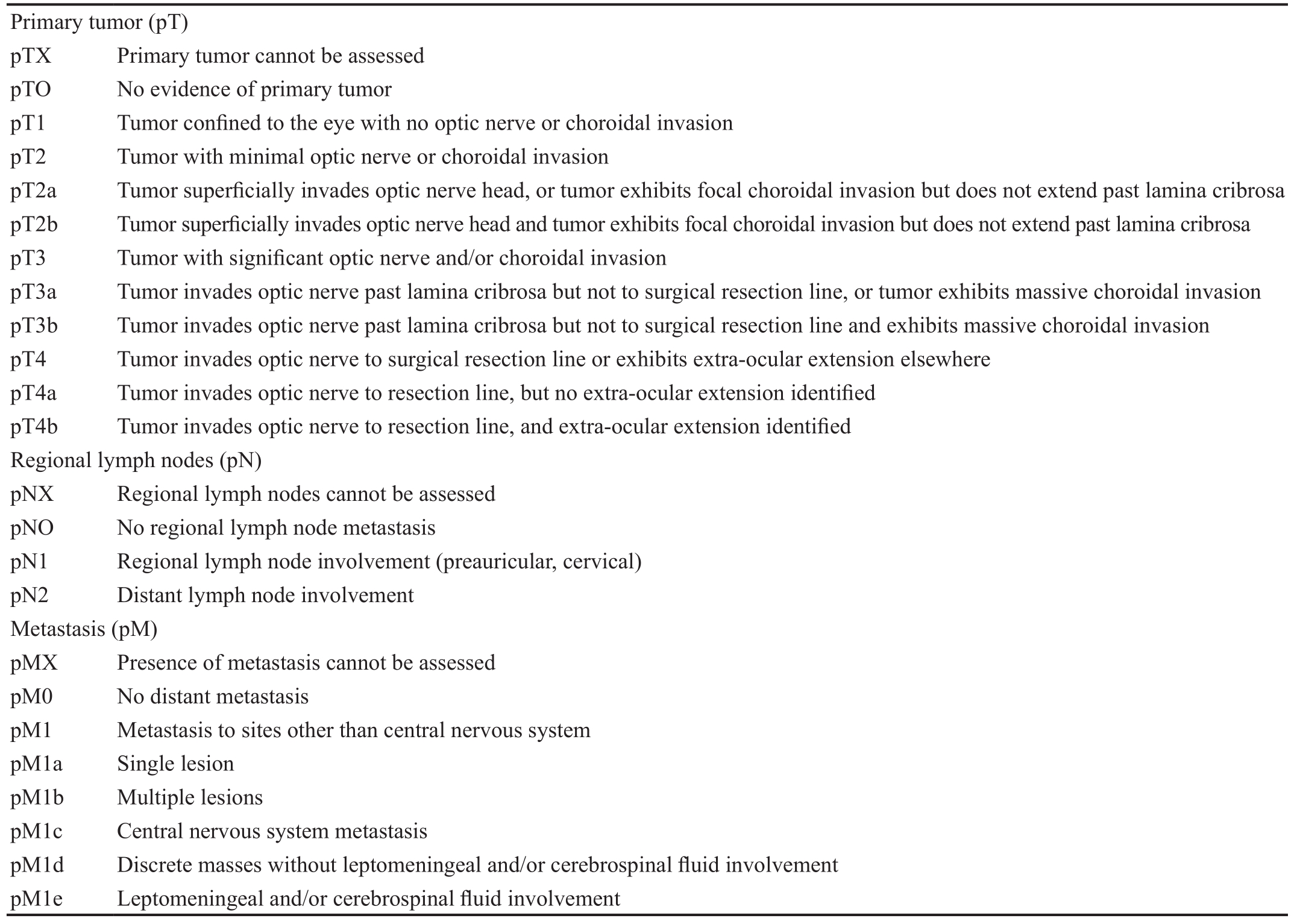
Table 1 AJCC pathological classi fication (pTNM)
?
Retinoblastoma is the most common intraocular malignancy of childhood and infancy accounting for 3% of all pediatrics cancers. It is caused by inactivation ofRB1genes commonly known as tumor suppressor gene[1]. Incidence of retinoblastoma ranges approximately worldwide at one case per 15 000-20 000 live births, which corresponds to about 9000 new cases every year[2]. There is no racial or gender predisposition. The sign of retinoblastoma is a white pupil, called leukocoria, strabismus, painful blind eye and loss of vision[3]. With the advancement in treatment and multidisciplinary approach, eye salvage is possible in group A through D intraocular tumours[4]. However, in group E tumours and group D (unilateral cases), enucleation always remains the choice of treatment. Higher incidences of histopathologic risk factors are reported from enucleated eyes from developing countries as opposed to developed countries[5].
Knudson’s putative tumor suppressor gene was cloned by Weinberg’s lab by studying genetic lesions and chromosomal aberrations in families with a history of retinoblastoma. This gene was the first human tumor suppressor gene to be cloned and it was named asRB1[6]. Initially, researchers considered thatRB1was important only for retinoblastoma susceptibility but Harbouret al[7]found that theRB1gene was also mutated in lung cancer.
India has the largest number of retinoblastoma cases with an estimated 1500 new cases annually[8]. It was the first tumour in which cancer genetics was revealed[9]. Cases with heritable retinoblastoma (48%) carry a germline mutation in theRB1gene and are likely to develop secondary cancers later in life like bone and soft tissue sarcomas, melanoma,brain tumours,etc.They have a 50% risk of transmitting their germlineRB1mutation to their offspring[10]. Cases with nonheritable retinoblastoma have normalRB1gene.Some of the retinoblastoma cases are caused byRB1gene mutation while others are caused by somatic ampli fication of theMYCNgene[11]. Recently, genetic laboratories have found that retinoblastoma may arise when theMYCNoncogene is amplified even in the presence of non-mutatedRB1genes.These cases are relatively rare, occuring in <3% of unilateral retinoblastoma cases[12]. Only 6% are familial while 94% are sporadic in newly diagnosed cases of retinoblastoma. All the cases of bilateral retinoblastoma involve germinal mutations[13].Almost 15% of unilateral sporadic retinoblastoma is caused by germinal mutations affecting only one eye while the 85% are sporadic.
Knudson proposed the two hit hypothesis in 1971[9]. Knudson stated that two chromosomal mutations are needed for developing retinoblastoma. The initial hit is a germinal mutation, which is inherited and is found in all cells in hereditary retinoblastoma. The second hit grows in the somatic retinal cells leading to the development of retinoblastoma.Therefore, hereditary cases are subjected to the development of non-ocular tumors such as osteosarcoma. In unilateral sporadic retinoblastoma, both the hits occur during the development of the retina and are somatic mutations[14]. Therefore, there is no risk of second nonocular tumors.
Genetic testing forRB1mutations and counselling of the patient can improve disease outcome and management. There are de finitive molecular tests which help in identifying children and their relatives who are at high risk for retinoblastoma,and need to be followed closely for the disease[15]. Recently,combinational approach of multiplex ligation-dependent probe ampli fication assay, deletion screening, direct sequencing, copy number gene dosage analysis and methylation assays provides mutational spectrum ofRB1gene mutation in retinoblastoma patients[16].
GrossThe gross appearance of retinoblastoma at cut section of the eye is somewhat variable, reflecting the stage of the disease at enucleation. The tumor has a white, encephaloid or brain-like appearance, with chalky areas of calcification and yellow necrotic areas[17]. The presence of calcium is often accentuated in eyes that have had prior radiotherapy or chemotherapy. Gross examination of the eyeball in the laboratory involves a total of 4 blocks. One block is the eyeball section with the optic nerve. Two blocks should contain the calottes. The fourth block consists of the resected margin of the optic nerve.
HistopathologicalOn microscopic examination, retinoblastoma reveals a tumor composed of small hyperchromatic cells with a high nuclear to cytoplasmic ratio with large areas of necrosis and multifocal area of calci fications. Tumour differentiation are categorized into well differentiated [>50% known as Homer-Wright (HW) rosettes] or poorly differentiated [<50% known as Flexner-Wintersteiner (FW) rosettes][18]. In 2014, new rosettes were found which were comparatively larger than FW and HW and has an unusal anterior segment involvement[19].Necrosis in the tumor is graded as none (<25%), mild (25%-50%), or extensive (>50%). Optic nerve invasion is graded as prelaminar, postlaminar and invasion of the resected margin.Postlaminar invasion is defined as tumour invasion beyond the lamina cribrosa of optic nerve[20]. Choroidal involvement is divided into focal invasion de fined as a tumour focus of less than 3 mm in any diameter (thickness or width) or massive invasion de fined as invasive focus of tumour measuring 3 mm or more in any diameter as per the Retinoblastoma Staging Working Group[21]. Artifactual seeding of tumour cells is seen at times in the sections which pose a problem to the pathologist. These are composed of small groups of tumour cells usually with many necrotic cells present within natural spaces of the eye. In contrast true tumour invasion comprises of solid nests of tumour cells with pushing or infiltrating borders without necrosis.
Histopathological Prognostic High Risk FactorsThe survival and management of high risk retinoblastoma has improved by identi fication of high-risk factors and appropriate adjuvant therapy. Histopathological high risk factors (HRFs)are evaluated and identified after enucleation for predicting metastasis. Prognostic factors like massive choroidal invasion,retrolaminar invasion and involvement of resected end of optic nerve, iris and ciliary body involvement, anterior chamber involvement, scleral and extrascleral involvement by tumour cells are associated with a greater risk of orbital recurrence and predictive of metastasis. There is a still debate regarding anterior chamber as a high-risk factor for retinoblastoma.Recently, Sreelakshmiet al[22]concluded in their study that anterior chamber seeds do not, by themselves, constitute an independent risk factor for metastasis in retinoblastoma. The reported incidences of HRFs are 7% to 56% for invasion of retrolaminar optic nerve and optic nerve to the transaction line; 12% to 42% for choroidal involvement; and 3% to 30%for scleral and extrascleral spread. Kashyapet al[23]described variousclinical features like older age at presentation,longer lag period, presence of hyphema, pseudohypopyon,staphyloma, and orbital cellulitis. These factors were associated with occurrence of HRFs and may be a useful indicator for considering adjuvant chemotherapy especially in developing countries. Also poorly differentiated retinoblastomas present at a later age and are associated with presence of multiple HRFs and necrosis[18]. Cases with presence of HRFs need systemic adjuvant chemotherapy which improves the survival of children at risk for metastatic disease[24]. Therefore,histopathologic HRFs can provide important basis for clinicians to determine treatment plan.
Pathological Tumor, Node, Metastasis Classi ficationTumor,node, metastasis (TNM) classification is developed by the American Joint Commission on Cancer (AJCC) and the Union International Control Cancer (Table 1)[25]. Retinoblastoma is the first cancer in which role of germline predisposition is recognised by incorporating stage category “H” into the AJCC classication[26]. Table 2 describes the AJCC 2017 8thedition tumor, node, metastasis, heritable trait (TNMH) clinical (c) and pathological (p) staging system which is known to be the first evidence-based system for predicting overall prognosis of both eye(s) and patients[27-28].
Ongoing studiesviahigher resolution genomic technologies,gene expression pro filing, direct gene sequencing, multiplexpolymerase chain reaction, mi-RNA microarray pro filing, nextgeneration sequencing (NGS), microsatellite analysis for loss of heterogeneity, andin-situhybridization for chromosomal aberrations will continue to facilitate our exploration into the molecular intricacies of retinoblastoma and results in newer therapeutic approaches.
The discovery of proto-oncogenes transformed our insight into mechanisms of cancer. More recent studies shows that retinoblastoma tumors may differ in the mutagenic pathway as some of retinoblastoma tumors are caused byRB1mutation[29-32]while some can also be initiated by ampli fication ofMYCNproto-oncogene.
Table 1 AJCC pathological classi fication (pTNM)
?
Singhet al[33]demonstrated prognostic significance of CDC25 phosphatases and polo-like kinases in retinoblastoma.They suggested that expression of CDC25B might be used as a potential prognostic marker in the pathogenesis of retinoblastoma and contribute to the development of the disease by causing genomic instability through deregulation of cell division. In their study, PLK1 was more frequently expressed and deregulated in poorly differentiated retinoblastoma tissue as compared to PLK3 protein that might serve as a poor prognostic marker in retinoblastoma[20].
Evasion of apoptosis is a hallmark of human cancers that leads to cancer development, progression and treatment resistance.The Bcl-2 family members are important regulators of the mitochondrial pathway of apoptosis. Bax and Bcl-2 are proteins that regulate programmed cell death and apoptosis. Recently,Singhet al[34]revealed higher expression of Bcl-2 in 66% of cases whereas Bax expression was found only in fewer cases(30%) of retinoblastoma tissue by immunohistochemistry,mRNA and Western blotting techniques. According to the author, differential expression of apoptotic regulatory proteins might represent poor response to patient outcome and have potential for tumor invasiveness.
Grottaet al[35]used a combined approach of next-generation sequencing (NGS) andRB1custom array-comparative genomic hybridization (aCGH) on a cohort of retinoblastoma patients. NGS andRB1custom aCGH have demonstrated to be an effective combined approach in order to optimize the overall diagnostic procedures of retinoblastoma. Devarajanet al[36]demonstrated for the first time that targeted next generation sequencing is an efficient approach for the identification of wide spectrum of pathogenic variants in retinoblastoma patients. Using this approach, an array of pathogenic variants including single nucleotide variants, InDels (small insertions/deletions) and copy number variations were detected in retinoblastoma patients. This comprehensive approach reduces the time and number of assays required for the detection of pathogenic variants by conventional methods which is sensitive (0.97) and ef ficient forRB1screening.
The application of genomics to the study of cancer is rapidly shifting toward the analysis of tissue samples to discover new biomarkers for early detection of cancers. Mitochondria have been implicated in tumor progression, cell differentiation, and apoptotic pathways. The identi fication of mitochondrial DNA mutations and its associated proteins as a biomarker has been used to help understand not only gene function but also the underlying molecular mechanisms of mitochondrial biology in retinoblastoma. This strategy relies on the hypothesis that if mutations in mtDNA cause physiological aberrationsspecifically in a particular tissue, the gene is more likely to be selectively expressed in that tissue. Currently, the role of mitochondria in retinoblastoma biology is still poorly understood.
Table 2 AJCC cTNMH retinoblastoma staging

?
Recently, Singhet al[37]have described and analyzed the morphological changes of mitochondria in retinoblastoma tumor by transmission electron microscopy. Poorly differentiated retinoblastoma cases showed fewer mitochondria,scant cytoplasm, disorganized organelles (mitochondria), and necrosis, whereas well-differentiated retinoblastomas had larger number of mitochondria and more organized organelles.Understanding the structural and functional characteristics of mitochondria in retinoblastoma might be essential for the design of future therapeutic strategies. They have also studied the expression of mitochondrial oxidative phosphorylation complexes in retinoblastoma tumor tissues. Among all the complexes, loss of mitochondrial complex I immunoexpression proved to be a useful independent prognostic biomarker to identify high-risk retinoblastoma patients[38]. Role of mitochondrial DNA and its protein biomarkers necessitate careful experimentation to adequately assess its contribution in retinoblastoma which might prove of diagnostic and prognostic value, and serve as a basis for the development of better long term therapeutic strategies. This provides an insight into molecular mechanisms of mitochondrial dysfunction, and also helps to find novel cancer biomarkers in retinoblastoma.
Sangeethaet al[39]investigated lipogenesis-dependent survival of retinoblastoma cancer cells and the associated molecular pathways in fatty acid synthase (FASN) silenced retinoblastoma cells and revealed that FASN silencing reduced the invading property of retinoblastoma cancer cells by scratch assay. Venkatesanet al[40]studied computational andin vitroinvestigation of miRNAs-gene regulation in retinoblastoma pathogenesis by anin silicoapproach. They concluded downregulation of miR-486-3p and miR-532-5p in primary retinoblastoma tissues, which might implicates their role in tumorigenesis.
Cytoplasmic expression of FOXO3a (transcription factor) has been found to be associated in pathogenesis of retinoblastoma.Relocation of FOXO3a from cytoplasmic to nucleus activates non-mutated retinoblastoma and might be a therapeutic target for retinoblastoma[41]. Reactive oxygen species and free radicals are associated with cancer development and its progression, which might suggest potential avenues of therapeutic intervention. Expression of NOX4 protein might be a source of reactive-oxygen species production in tumor cells,leading to oxidative stress and associated with less overall survival rate in retinoblastoma[42].
Sirt1 (Sirtuin1) is the most important protein among all the sirtuins as it involves multiple factors that are highly relevant to cancer. Batraet al[43]found high expression of Sirt1 in retinoblastoma tumors but it was not associated with any high-risk histopathological factors. Similarly, Sirt2 and Sirt6 were also expressed in tumor cells along with various normal structures of the remaining ocular tissues[44]. Apart from the expression studies, mass spectrometry-based quantitative proteomic approach was also implemented to identify differentially expressed proteins in which mitochondrial dysfunction and lipid metabolism pathways found to be deregulated in retinoblastoma tumor[45].
Recently, an elevated expression of PDK1 protein levels has been validated in retinoblastoma tumors especially in vitreous seeds and hypoxic regions. Inhibition ofPDK1gene in retinoblastoma cell lines demonstrated reduced cell growth and increased apoptosis, which might be a potential future therapeutic target in retinoblastoma[46].
Current treatment strategies contribute signi ficantly to vision salvage in patients harbouring intraocular diseases, and overall survival rates in patients with extraocular disease.However therapies such as chemotherapy, brachytherapy and plaque therapy do result in significant morbidity. A better understanding of pathobiology of retinoblastoma may lead to better outcome in therapies with less long-term morbidity and prevent onset of secondary cancers. Translational research has shown the ef ficacy of adjuvant treatment in animal model and retinoblastoma cell lines leading to new insights into the development of retinoblastoma. Future trends in retinoblastoma treatment will focus on the local delivery of optimally-timed therapeutic agents which will act synergistically in the control of retinoblastoma tumors.
Conflicts of Interest: Singh L, None; Kashyap S, None.
1 Dunn JM, Phillips RA, Becker AJ, Gallie BL. Identi fication of germline and somatic mutations affecting the retinoblastoma gene.Science1998;241(4874):1797-1800.
2 Kivela T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death.Br J Ophthalmol2009;93(9):1129-1131.
3 Abramson DH, Frank CM, Susman M, Whalen MP, Dunkel IJ, Boyd NW 3rd. Presenting signs of retinoblastoma.J Pediatr1998;132(3 Pt 1):505-508.
4 Yousef YA, Hajja Y, Nawaiseh I, Mehyar M, Sultan I, Deebajah R,Rawashdeh K, Khurma S, Jaradat I, Al-Hussaini M. A histopathologic analysis of 50 eyes primarily enucleated for retinoblastoma in a tertiary cancer center in Jordan.Turk Patoloji Derg2014;30(3):171-177.
5 Vemuganti G, Honavar S, John R. Clinicopathological profile of retinoblastoma patients in Asian Indians.Invest Ophthalmol Vis Sci2000;41(S):790.
6 Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM,Albert DM, Dryja TP. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma.Nature1986;323(6089):643-646.
7 Harbour JW, Lai SL, Whang-Peng J, Gazdar AF, Minna JD, Kaye FJ.Abnormalities in structure and expression of the human retinoblastoma gene in SCLC.Science1988;241(4863):353-357.
8 Dimaras H, Kimani K, Dimba EA, Gronsdahl P, White A, Chan HS,Gallie BL. Retinoblastoma.Lancet2012;379(9824):1436-1446.
9 Dimaras H. Retinoblastoma genetics in India: from research to implementation.Indian J Ophthalmol2015;63(3):219-226.
10 Garber JE, Offit K. Hereditary cancer predisposition syndromes.J Clin Oncol2005;23(2):276-292.
11 Rushlow DE, Mol BM, Kennett JY,et al. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies.Lancet Oncol2013;14(4):327-334.
12 Fabian ID, Rosser E, Sagoo MS. Epidemiological and genetic considerations in retinoblastoma.Community Eye Health2018;31(101):29-30.
13 Shields JA, Shields CL.Genetics of retinoblastoma.In: Shields JA,Shields CL, eds. Intraocular tumors: a text and Atlas. Philadelphia, Pa:WB Saunders;1992:333-340.
14 Ray A, Gombos DS, Vats TS. Retinoblastoma: an overview.Indian J Pediatr2012;79(7):916-921.
15 Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP,Plon SE, Chévez-Barrios P. Screening children at risk for retinoblastoma:consensus report from the American association of ophthalmic oncologists and pathologists.Ophthalmology2018;125(3):453-458.
16 Tomar S, Sethi R, Sundar G, Quah TC, Quah BL, Lai PS. Mutation spectrum of RB1 mutations in retinoblastoma cases from Singapore with implications for genetic management and counselling.PLoS One2017;12(6):e0178776.
17 Eagle RC Tr.Eye pathology: an Atlas and text.2ndedition. Philadelphia:Wolters Kluwer/Lippincott Williams & Wilkins;2011.
18 Kashyap S, Sethi S, Meel R, Pushker N, Sen S, Bajaj MS, Chandra M, Ghose S. A histopathologic analysis of eyes primarily enucleated for advanced intraocular retinoblastoma from a developing country.Arch Pathol Lab Med2012;136(2):190-193.
19 Das D, Bhattacharjee K, Barthakur SS, Tahiliani PS, Deka P,Bhattacharjee H, Deka A, Paul R. A new rosette in retinoblastoma.Indian J Ophthalmol2014;62(5):638-641.
20 Singh L, Pushker N, Sen S, Singh MK, Chauhan FA, Kashyap S.Prognostic signi ficance of polo-like kinases in retinoblastoma: correlation with patient outcome, clinical and histopathological parameters.Clin Exp Ophthalmol2015;43(6):550-557.
21 Sastre X, Chantada GL, Doz F, Wilson MW, de Davila MT,Rodríguez-Galindo C, Chintagumpala M, Chévez-Barrios P; International Retinoblastoma Staging Working Group. Proceedings of the consensus meetings from the International Retinoblastoma Staging Working Group on the pathology guidelines for the examination of enucleated eyes and evaluation of prognostic risk factors in retinoblastoma.Arch Pathol Lab Med2009;133(8):1199-1202.
22 Sreelakshmi KV, Chandra A, Krishnakumar S, Natarajan V, Khetan V. Anterior chamber invasion in retinoblastoma: not an indication for adjuvant chemotherapy.Invest Ophthalmol Vis Sci2017;58(11):4654-4661.
23 Kashyap S, Meel R, Pushker N, Sen S, Bakhshi S, Sreenivas V, Sethi S, Chawla B, Ghose S. Clinical predictors of high risk histopathology in retinoblastoma.Pediatr Blood Cancer2012;58(3):356-361.
24 Eagle RC Jr. High-risk features and tumor differentiation in retinoblastoma: a retrospective histopathologic study.Arch Pathol Lab Med2009;133(8):1203-1209.
25 Finger PT, Harbour JW, Murphree AL.Retinoblastoma.In: Edge DR,Byrd DR, Compton CC, editors. AJCC cancer staging manual 2009, vol. 7.New York, NY: Springer.
26 AlAli A, Kletke S, Gallie B, Lam WC. Retinoblastoma for pediatric ophthalmologists.Asia Pac J Ophthalmol (Phila)2018;7(3):160-168.
27 Mallipatna A, Gallie BL, Chévez-Barrios P.Retinoblastoma.In: Amin MB, Edge SB, Greene FL, eds. AJCC Cancer Staging Manual. 8th ed.New York, NY: Springer;2017:819-831.
28 Fabian ID, Onadim Z, Karaa E, Duncan C, Chowdhury T, Scheimberg I, Ohnuma S, Reddy MA, Sagoo MS. The management of retinoblastoma.Oncogene2018;37(12):1551-1560.
29 Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma.Proc Natl Acad Sci U S A1971;68(4):820-823.
30 Comings DE. A general theory of carcinogenesis.Proc Natl Acad Sci U S A1973;70(12):3324-3328.
31 Dryja TP, Friend S, Weinberg RA. Genetic sequences that predispose to retinoblastoma and osteosarcoma.Symp Fundam Cancer Res1986;39:115-119.
32 Khetan V, Gupta A, Gopal L. Retinoblastoma: recent trends a mini review based on published literature.Oman J Ophthalmol2011;4(3):108-115.
33 Singh L, Pushker N, Sen S, Singh MK, Bakhshi S, Chawla B, Kashyap S. Expression of CDC25A and CDC25B phosphatase proteins in human retinoblastoma and its correlation with clinicopathological parameters.Br J Ophthalmol2015;99(4):457-463.
34 Singh L, Pushker N, Saini N, Sen S, Sharma A, Bakhshi S, Chawla B,Kashyap S. Expression of pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins in human retinoblastoma.Clin Exp Ophthalmol2015;43(3):259-267.
35 Grotta S, D’Elia G, Scavelli R,et al. Advantages of a next generation sequencing targeted approach for the molecular diagnosis of retinoblastoma.BMC Cancer2015;15:841.
36 Devarajan B, Prakash L, Kannan TR, Abraham AA, Kim U,Muthukkaruppan V, Vanniarajan A. Targeted next generation sequencing of RB1 gene for the molecular diagnosis of Retinoblastoma.BMC Cancer2015;15:320.
37 Singh L, Nag TC, Kashyap S. Ultrastructural changes of mitochondria in human retinoblastoma: correlation with tumor differentiation and invasiveness.Tumour Biol2016;37(5):5797-5803.
38 Singh L, Saini N, Bakhshi S, Pushker N, Sen S, Sharma A, Kaur J, Kashyap S. Prognostic significance of mitochondrial oxidative phosphorylation complexes: therapeutic target in the treatment of retinoblastoma.Mitochondrion2015;23:55-63.
39 Sangeetha M, Deepa PR, Rishi P, Khetan V, Krishnakumar S.Global gene deregulations in FASN silenced retinoblastoma cancer cells: molecular and clinico-pathological correlations.J Cell Biochem2015;116(11):2676-2694.
40 Venkatesan N, Deepa PR, Khetan V, Krishnakumar S. Computational and in vitro investigation of miRNA-gene regulations in retinoblastoma pathogenesis: miRNA mimics strategy.Bioinform Biol Insights2015;9:89-101.
41 Batra A, Kashyap S, Singh L, Bakhshi S. Expression of FOXO3a and correlation with histopathologic features in retinoblastoma.Appl Immunohistochem Mol Morphol2017;25(2):95-99.
42 Singh L, Saini N, Pushker N, Sen S, Sharma A, Kashyap S. Prognostic significance of NADPH oxidase-4 as an indicator of reactive oxygen species stress in human retinoblastoma.Int J Clin Oncol2016;21(4):651-657.
43 Batra A, Kashyap S, Singh L, Bakhshi S. Sirtuin1 expression and correlation with histopathological features in retinoblastoma.Ocul Oncol Pathol2015;2(2):86-90.
44 Orellana ME, Quezada C, Maloney SC, Antecka E, Balazsi M, Burnier MN Jr. Expression of SIRT2 and SIRT6 in retinoblastoma.Ophthalmic Res2015;53(2):100-108.
45 Danda R, Ganapathy K, Sathe G, Madugundu AK, Ramachandran S, Krishnan UM, Khetan V, Rishi P, Keshava Prasad TS, Pandey A, Krishnakumar S, Gowda H, Elchuri SV. Proteomic profiling of retinoblastoma by high resolution mass spectrometry.Clin Proteomics2016;13:29.
46 Sradhanjali S,Tripathy D, Rath S, Mittal R, Reddy MM. Overexpression of pyruvate dehydrogenase kinase 1 in retinoblastoma: a potential therapeutic opportunity for targeting vitreous seeds and hypoxic regions.PLoS One2017;12(5):e0177744.