Biomarker identification of thyroid associated ophthalmopathy using microarray data
Hong-Bin Yang 1 , Jie Jiang 1 , Lu-Lu Li 2 , Huang-Qiang Yang 2 , Xiao-Yu Zhang 2
1 Department of Ophthalmology, the First Affiliated Hospital of Harbin Medical University, Harbin 150080, Heilongjiang Province, China
2 Department of Neurosurgery, the Second Affiliated Hospital of Harbin Medical University, Harbin 150080, Heilongjiang Province, China
· KEYWORDS: microarray; thyroid associated ophthalmopathy;cell cycle; biomarker
INTRODUCTION
G raves’ disease is a type of autoimmune problem that causes the thyroid gland to produce too much thyroid hormone, which is called hyperthyroidism. Whereas,thyroid associated ophthalmopathy (TAO), also called Graves’ orbitopathy, which is closely correlated with Graves’hyperthyroidism (GH), is normally considered as an inflammatory autoimmune eye disorder [1-2] . Annually, it is reported that 16 of per 100 000 females and 2.9 of per 100 000 males are suffered from TAO [3] . The current therapeutic strategies were through treating with corticosteroids, external beam radiation or surgery, however, the occurrence of this disease could not be prevented. Previously, the thyrotropin receptor (TSHR)was demonstrated be closely associated with TAO, and it was hypothesized that the progression of ophthalmopathy is the autoimmunity against TSHR in the orbital preadipocyte and extraocular muscle fibroblasts [4-5] . However, due to the inadequate interpretation for eye muscle swelling and the lack of correlation between TSHR antibody level and eye signs,more appropriate theories are explored by other investigations.Some researchers prefer to stress the significant role of autoimmunity against orbital connective tissue (OCT) [6] and eye muscle antigens [7] . Among them, the calcium binding protein calsequestrin was highly over-expressed in eye muscle and was proposed as the causative factor for the orbital skeletal muscle inflammation in TAO [8] . Further investigation conducted by Lahooti et al [9] suggested that calsequestrin might serve as a candidate in the pathogenesis of TAO. In addition,they also explored the role of the OCT antigen collagen XIII and indicated the serum antibodies against collagen XIII may contribute to the congestive ophthalmopathy [9] . Multiple inflammation relating biomarkers including ICAM-1 , VCAM-1 and TIMP-1 have been linked to autoimmune thyroid disorders [10] but only a handful of genetic factors such as PTPN22 , TSHR , CTLA and CD40 as well as AFF3 and CD226 were involved in the TAO biomarker establishment [11] .
Recently, a study used microarray analysis to identify differentially expressed genes (DEGs) between GH (thyroid Graves’ patients without ophthalmopathy) and TAO. Though the study screened 295 significant DEGs, their emphasis was on the correlation between the most up-regulated cardiac calsequestrin gene ( CASQ2 ) and TAO [12] , no more additional relevant genes were elaborated. Therefore, we re-analyzed the microarray data to further explore crucial DEGs between TAO and GH patients without ophthalmopathy, followed by the functional enrichment analysis for the selected DEGs.Moreover, a protein-protein interaction (PPI) network was constructed, from which specific modules are extracted to obtain more exact correlations between the DEGs, attempting to uncover the pathogenesis of TAO and provide potential biomarkers for the prognosis and therapeutic methods of TAO.
SUBJECTS AND METHODS
Microarray Data The expression profile GSE9340, which was deposited in the GEO (Gene Expression Omnibus; http://www.ncbi.nlm.nih.gov/geo) database by Wescombe et al [12] ,included 18 thyroid tissue samples from 10 TAO patients(TAO group) and 8 GH patients without ophthalmopathy(control group). The platform was HumanRef-8 v2 Expression BeadChip.
Ethics Approval and Consent to Participate This study was approved by Ethics Committee of the First Affiliated Hospital of Harbin Medical University and the Second Affiliated Hospital of Harbin Medical University.
Data Preprocessing and the Identification of Differentially Expressed Genes Based on the profiling files, probes were converted to the gene symbols. The probe not corresponding to gene symbols were removed, and when multiple probes corresponded to a single gene, the average level of probes was calculated as the expression value of the specific gene. The raw data were normalized using median method of preprocess Core package [13] . After the data preprocessing, package limma(Linear Models for Microarray Analysis) of Bioconductor R(http://www.bioconductor.org/packages/release/bioc/html/limma.html) [14] was used to identify the DEGs between TAO and control groups, basing on t -test method. The genes met with the criteria of P value <0.05 and |log 2 (fold change)|≥1.5 were considered as DEGs.
Functional Enrichment Analysis The selected DEGs were mapped to gene ontology (GO; http: //www. geneontology.org/) database to explore their potential process in terms of biological process (BP), molecular function (MF) and cellular component (CC), using the tool of DAVID (Database for Annotation, Visualization and Integration Discovery; http://david.abcc.Ncifcrf.gov/) [15] . Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/pathway.html) enrichment analysis was performed to identify the pathways in which these DEGs might involve.The threshold based on hypergeometric test for significant GO terms and pathways were P <0.05.
Protein-protein Interaction Network Construction Considering that protein encoding by these selected DEGs might exert their functions combining with other proteins, the DEGs were mapped to STRING (Search Tool for the Retrieval of Interacting Genes; http://string-db.org/) [16] database to identify the potential interactions between them and other genes from protein level. The interaction with a combined score >0.7 was screened to establish the PPI network,visualizing by Cytoscape software [17] .
Furthermore, the ClusterOne [18] , a Cytoscape software plugin,was employed to explore the functional modules of the network and subsequently, followed by the GO enrichment analysis for significant modules, using the BiNGO plugin [19] .The P value was adjusted to false discover rate (FDR) and the cut-off value for significant BP terms was FDR<0.05.
Prediction of Transcription Factor and Other Related Genes Combining the information of transcription factors(TFs) with their targets in University of California Santa Cruz(UCSC) Genome Browser database (http://genome. ucsc.edu) [20] , the TFs among these DEGs were predicted as well as the corresponding targets. Subsequently, Comparative Toxicogenomics Database (CTD; http://ctdbase. org/) [21] was used to identify TAO-related genes among our selected DEGs.The database collects disease-related genes which have been validated by experiment or been reported by literature.
RESULTS
Identification of Differentially Expressed Genes Between Thyroid Associated Ophthalmopathy and Control Groups Data after normalization exhibited a uniform distribution of the mean value, indicating a favorable normalization that the data were eligible to identify the DEGs. According to the aforementioned criteria, a total of 433 up-regulated and 428 down-regulated DEGs were screened.
Over-represented Processes and Pathways by Enrichment Analysis With the predefined criteria, correlated processes and pathways which the DEGs might involve in were filtered out.The up-regulated DEGs were predominantly enriched in GO terms such as generation of precursor metabolites and energy(GO:0006091), oxidation reduction (GO:0055114), glycolysis(GO:0006096), mitochondrion (GO:0005739) and 4 iron, 4 sulfur cluster binding (GO:0051539); while the down-regulated DEGs were significantly correlated with cell cycle related processes (GO:0051301, GO:0007049 and GO:0022403) and regulation of transcription (GO:0045449) (Table 1). KEGG enrichment analysis indicated that the over-presented pathways for the up-regulated DEGs were fructose and mannose metabolism, protein export and citrate cycle, whereas the down-regulated DEGs were mainly enriched in systemic lupus erythematosus, oocyte meiosis and progesterone-mediated oocyte maturation (Table 2).
Constructed Protein-protein Interaction Network of the Differentially Expressed Genes and the Module Analysis By mapping the protein information in STRING database, a PPI network was established for the protein products of the DEGs, embracing 696 interactions and 339 nodes. A protein in the PPI network was defined as a “node” and the “degree” of the node referred to the interaction number of that particular protein. The most highly connected nodes with high degree were deemed as the “hubs” of the network. The remarkable hubs in the PPI network were presented in Figure 1, embracing BUB1B (degree=35), CDC20 (degree=33), CDCA8(degree=26), PRC1 (degree=24), CENPA (degree=24), CCNB2(degree=24), TPX2 (degree=23), CDCA5 (degree=23), KIF23(degree=23) and SPC25 (degree=23).
Using the ClusterOne plugin, a significant module was extracted from the PPI network, involving 236 interactions and 27 nodes, in which the nodes with high degrees were highlighted including SPC25 (degree=23), TPX2 (degree=23),CENPA (degree=23), KIF23 (degree=22), PRC1 (degree=22),CDCA5 (degree=21), HMMR (degree=21), KIF20A(degree=21), RAD51AP1 (degree=21) and MKI67 (degree=19)(Figure 2). In addition, there were some interactions between these hub protein pairs, such as TPX and KIF23 or MKI67 or PRC1, MKI67 and PRC1 or KIF23 or CDCA5, as well as CDCA5 and KIF23. The functional enrichment for genes in the module was performed by the BinGO plugin. As presented in Table 3, genes which encoded the predominant nodes in the module (such as KIF23 , TPX2 , MKI67 , PRC1 and CDCA5 )were mainly correlated with the processes related to cell division and cell cycle.
Predicted Transcription Factors As a result, a total of 88 TFs involving 4700 targeting interactions were predicted through the comparison with the TFs in UCSC database.Among them, four TFs were encoded by DEGs such as GTF2F1 , SMC3 , USF1 and ZNF263 , in which SMC3 was upregulated, while others were down-regulated. The targets of the four TFs were revealed in Figure 3.
Thyroid Associated Ophthalmopathy Related Genes We obtained 1406 genes associated with TAO from CTD database,including 42 DEGs in the present study (Table 4). In other words, these DEGs were prominent in TAO pathogenesis.Notably, the down-regulated MKI67 with a high degree of 19,which was a hub protein in the module, was highly associated with TAO.
Table 1 Significant enriched GO terms of up- and down-regulated DEGs in thyroid-associated ophthalmopathy (top 5 ranked by P value)
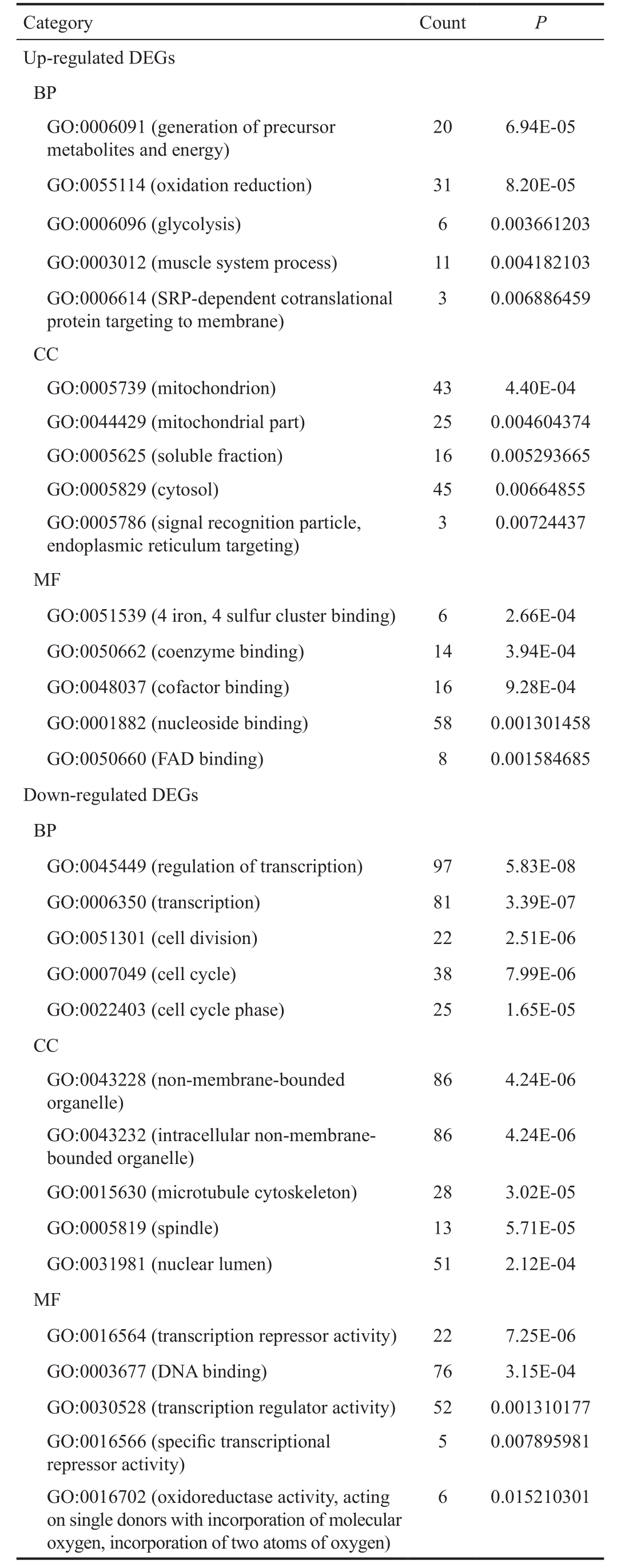
GO: Gene ontology; DEG: Differentially expressed genes; Count:Number of the DEGs in a specific term; BP: Biological process; MF:Molecular function; CC: Cellular components.
Category Count P Up-regulated DEGs BP GO:0006091 (generation of precursor metabolites and energy)20 6.94E-05 GO:0055114 (oxidation reduction) 31 8.20E-05 GO:0006096 (glycolysis) 6 0.003661203 GO:0003012 (muscle system process) 11 0.004182103 GO:0006614 (SRP-dependent cotranslational protein targeting to membrane)3 0.006886459 CC GO:0005739 (mitochondrion) 43 4.40E-04 GO:0044429 (mitochondrial part) 25 0.004604374 GO:0005625 (soluble fraction) 16 0.005293665 GO:0005829 (cytosol) 45 0.00664855 GO:0005786 (signal recognition particle,endoplasmic reticulum targeting)3 0.00724437 MF GO:0051539 (4 iron, 4 sulfur cluster binding) 6 2.66E-04 GO:0050662 (coenzyme binding) 14 3.94E-04 GO:0048037 (cofactor binding) 16 9.28E-04 GO:0001882 (nucleoside binding) 58 0.001301458 GO:0050660 (FAD binding) 8 0.001584685 Down-regulated DEGs BP GO:0045449 (regulation of transcription) 97 5.83E-08 GO:0006350 (transcription) 81 3.39E-07 GO:0051301 (cell division) 22 2.51E-06 GO:0007049 (cell cycle) 38 7.99E-06 GO:0022403 (cell cycle phase) 25 1.65E-05 CC GO:0043228 (non-membrane-bounded organelle)86 4.24E-06 GO:0043232 (intracellular non-membranebounded organelle)86 4.24E-06 GO:0015630 (microtubule cytoskeleton) 28 3.02E-05 GO:0005819 (spindle) 13 5.71E-05 GO:0031981 (nuclear lumen) 51 2.12E-04 MF GO:0016564 (transcription repressor activity) 22 7.25E-06 GO:0003677 (DNA binding) 76 3.15E-04 GO:0030528 (transcription regulator activity) 52 0.001310177 GO:0016566 (specific transcriptional repressor activity)5 0.007895981 GO:0016702 (oxidoreductase activity, acting on single donors with incorporation of molecular oxygen, incorporation of two atoms of oxygen)6 0.015210301
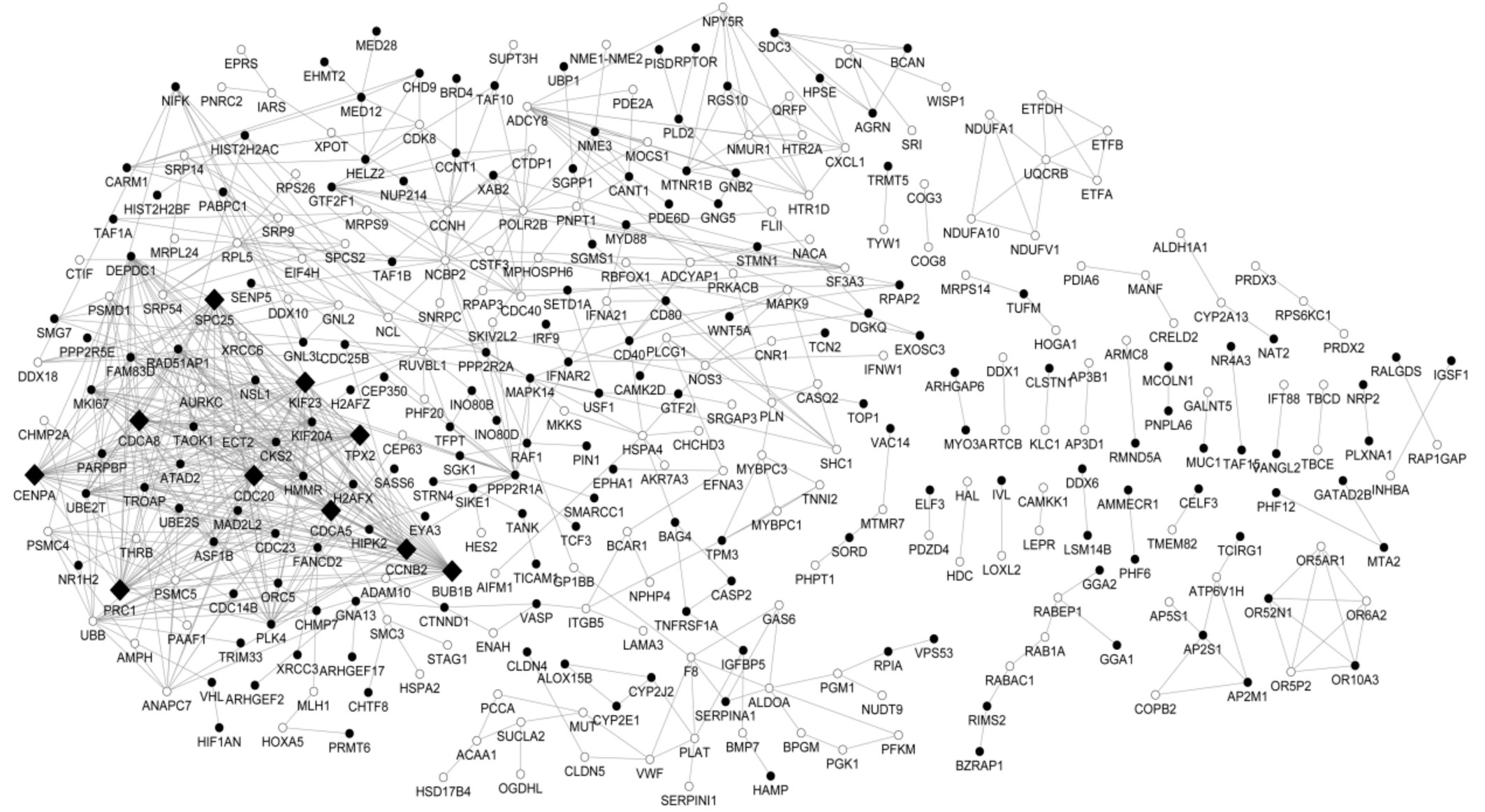
Figure 1 Protein-protein interaction network of the DEGs The white round represents proteins encoded by up-regulated DEGs and the black round represents proteins encoded by down-regulated DEGs. The protein products in the networks serve as the “nodes”, and the undirected link denotes each pairwise interaction. The most highly connected nodes with high degree were deemed as the “hubs”. In the network, the black rhombus indicates hub proteins.
Table 2 Significant enriched pathways for up- and down-regulated DEGs
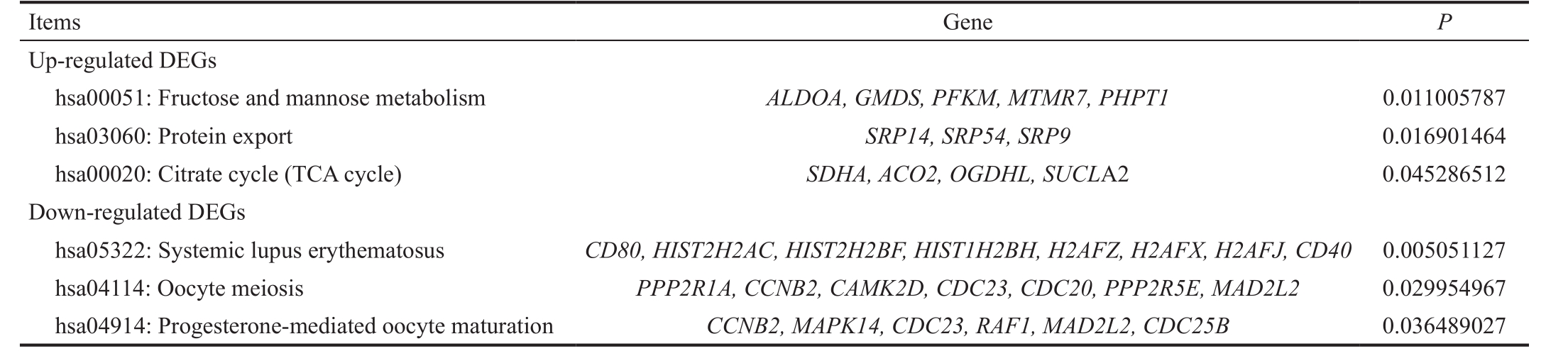
DEG: Differentially expressed genes.
Gene P Up-regulated DEGs hsa00051: Fructose and mannose metabolism ALDOA, GMDS, PFKM, MTMR7, PHPT1 0.011005787 hsa03060: Protein export SRP14, SRP54, SRP9 0.016901464 hsa00020: Citrate cycle (TCA cycle) SDHA, ACO2, OGDHL, SUCL A2 0.045286512 Down-regulated DEGs hsa05322: Systemic lupus erythematosus CD80, HIST2H2AC, HIST2H2BF, HIST1H2BH, H2AFZ, H2AFX, H2AFJ, CD40 0.005051127 hsa04114: Oocyte meiosis PPP2R1A, CCNB2, CAMK2D, CDC23, CDC20, PPP2R5E, MAD2L2 0.029954967 hsa04914: Progesterone-mediated oocyte maturation CCNB2, MAPK14, CDC23, RAF1, MAD2L2, CDC25B 0.036489027 Items
Table 3 Enrichment analysis for genes in the module
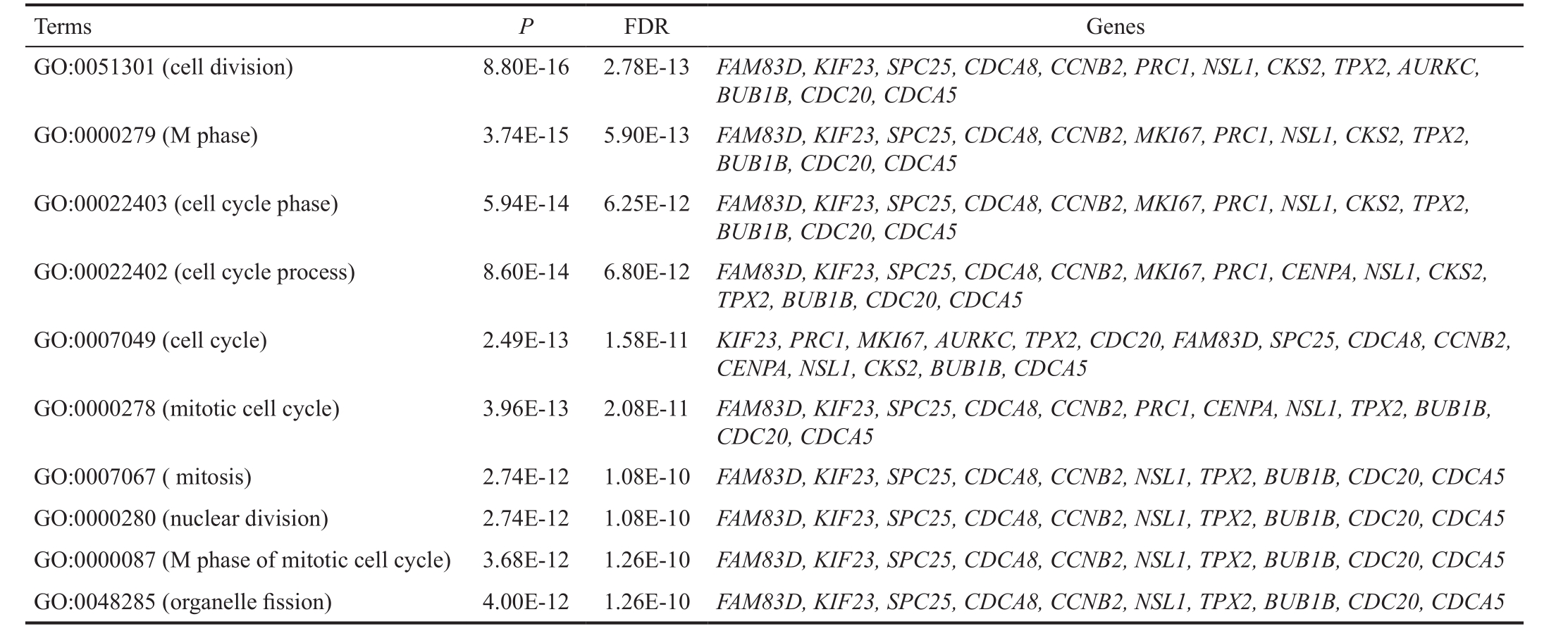
GO: Gene ontology; FDR: False discovery rate.
Terms P FDR Genes GO:0051301 (cell division) 8.80E-16 2.78E-13 FAM83D, KIF23, SPC25, CDCA8, CCNB2, PRC1, NSL1, CKS2, TPX2, AURKC, BUB1B, CDC20, CDCA5 GO:0000279 (M phase) 3.74E-15 5.90E-13 FAM83D, KIF23, SPC25, CDCA8, CCNB2, MKI67, PRC1, NSL1, CKS2, TPX2, BUB1B, CDC20, CDCA5 GO:00022403 (cell cycle phase) 5.94E-14 6.25E-12 FAM83D, KIF23, SPC25, CDCA8, CCNB2, MKI67, PRC1, NSL1, CKS2, TPX2, BUB1B, CDC20, CDCA5 GO:00022402 (cell cycle process) 8.60E-14 6.80E-12 FAM83D, KIF23, SPC25, CDCA8, CCNB2, MKI67, PRC1, CENPA, NSL1, CKS2, TPX2, BUB1B, CDC20, CDCA5 GO:0007049 (cell cycle) 2.49E-13 1.58E-11 KIF23, PRC1, MKI67, AURKC, TPX2, CDC20, FAM83D, SPC25, CDCA8, CCNB2, CENPA, NSL1, CKS2, BUB1B, CDCA5 GO:0000278 (mitotic cell cycle) 3.96E-13 2.08E-11 FAM83D, KIF23, SPC25, CDCA8, CCNB2, PRC1, CENPA, NSL1, TPX2, BUB1B, CDC20, CDCA5 GO:0007067 ( mitosis) 2.74E-12 1.08E-10 FAM83D, KIF23, SPC25, CDCA8, CCNB2, NSL1, TPX2, BUB1B, CDC20, CDCA5 GO:0000280 (nuclear division) 2.74E-12 1.08E-10 FAM83D, KIF23, SPC25, CDCA8, CCNB2, NSL1, TPX2, BUB1B, CDC20, CDCA5 GO:0000087 (M phase of mitotic cell cycle) 3.68E-12 1.26E-10 FAM83D, KIF23, SPC25, CDCA8, CCNB2, NSL1, TPX2, BUB1B, CDC20, CDCA5 GO:0048285 (organelle fission) 4.00E-12 1.26E-10 FAM83D, KIF23, SPC25, CDCA8, CCNB2, NSL1, TPX2, BUB1B, CDC20, CDCA5

Figure 2 Module of the DEGs The white round represents proteins encoded by up-regulated DEGs and the black represents proteins encoded by down-regulated DEGs. The protein products in the networks serve as the ’nodes’, and the undirected link denotes each pairwise interaction.
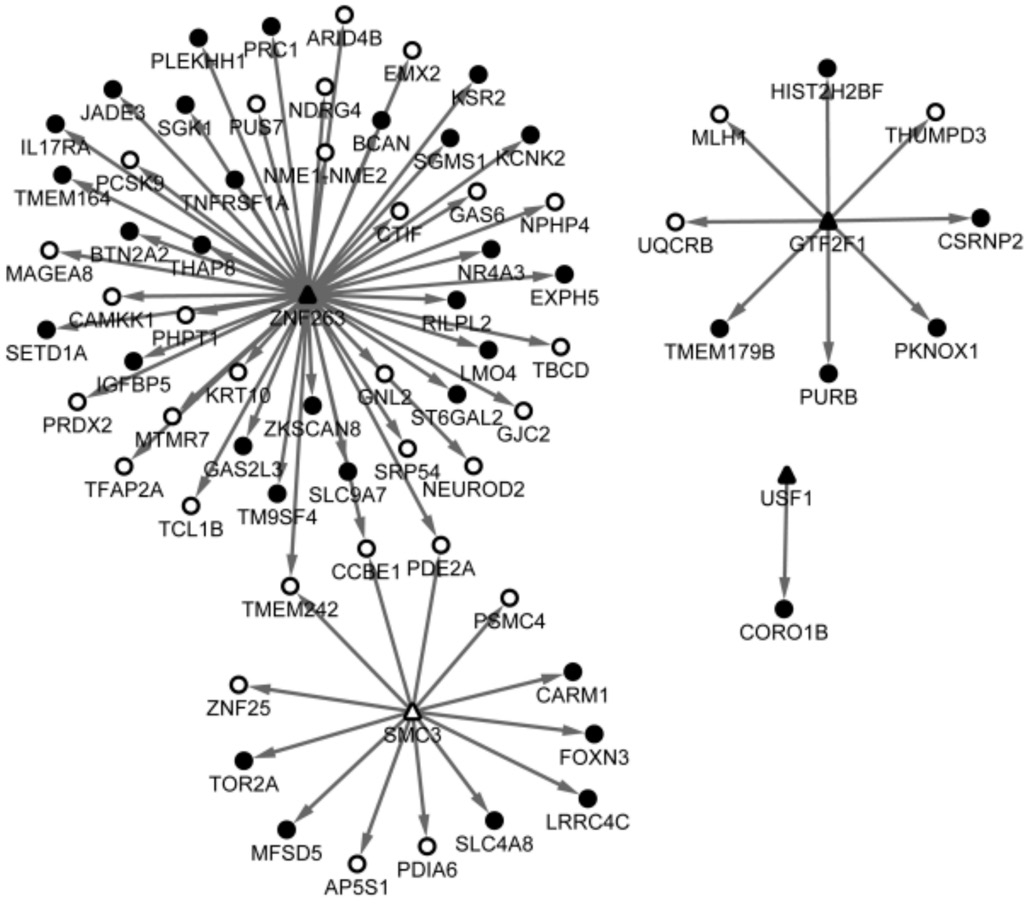
Figure 3 Predicted targets of the TFs The triangle represents TF and the round represents the targets, in which the white denotes upregulated and the black denotes down-regulated. The arrow indicates the relationships between TFs and the targets.
DISCUSSION
As a result, a total of 861 DEGs (433 were up-regulated and 428 were down-regulated) between TAO and control groups were identified. Crucial nodes in the PPI network included TPX2, CDCA5, PRC1, KIF23 and MKI67, which were also highlighted in the module of the network and all enriched in cell cycle related processes. Additionally, MKI67 was highly correlated with TAO. Besides, the DEGs of GTF2F1 , SMC3 ,USF1 and ZNF263 were predicted as TFs.
TPX2 (Targeting Protein for the Xenopuskinesinxklp2) is essential for normal spindle assembly and meiotic divisions,and also is cell cycle-mediated in somatic cells [22] . During the stage of meiotic divisions in mouse oocytes, TPX2 is acentral target of Ran, the active form of which (Ran-GTP) was essential for mitotic spindles organization [23] . By interacting with the Kinesin-5 motor Eg5, another spindle-associated protein, TPX2 was demonstrated to facilitate to the localization of Eg5 to spindle microtubules with the bacterial artificial chromosome method [24] . CDCA5 (cell division cycle associated 5) is another cell cycle associated gene which mainly serves as a regulator of sister chromatid cohesion in mitosis stabilizing cohesin complex. The human breast cancer-associated fibroblast genes including CDCA5 and TRIP13 (thyroid hormone receptor interactor 13), which played significant roles in chromosome recombination and chromosome structure development during meiosis, were enriched in cell cycle BP function [25] . Though there were rare evidence that validated the relationships between TPX2 and CDCA5 especially in TAO disease, a study using genome-scale RNAi profiling technique indicated that both TPX2 and CDCA5 (KIF20A)were associated with cell division phenotypes [26] . In the present study, as two vital nodes in the module of the network, CDCA5 and TPX2 were both enriched in mitotic cell cycle function,and notably, CDCA5 was linked to TPX2, providing a hint that the dysfunction in mitotic cell cycle process, which might be mediated by the interaction of TPX2 and CDCA5 , might play important roles in the TAO associated events.
Table 4 The common DEGs (degree>2) based on the genes associated with TAO from CTD database

Genes Degree MKI67 19 UBB 17 PPP2R1A 14 CXCL1 7 F8 6 UQCRB 6 CD40 6 PLAT 5 DCN 5 VWF 4 IFNAR 4 AGRN 4 TPM3 4 NOS3 4 NDUFA10 3 NDUFA1 3 NDUFV1 3 SUCLA2 3 ETFDH 3 ETFA 3 MUT 3 ACAA1 3 ETFB 3
PRC1 and KIF23 were another two remarkable nodes as revealed in the module. The protein encoded by PRC1 is a central-spindle matrix protein and involved in cytokinesis [27-28] .The absence of PRC1 could result in the disorganization of motor protein. Besides, it was convinced that PRC1 was required in the MKlp1 (the kinesin-6 family motor protein)mediated targeting to the central spindle [29] . As a kinesin-like protein family member, KIF23 is also involved in cell division.Notably, the protein KIF23 serves as a part of the central spindle, which exhibited at the spindle midzone as a complex of PRC1 and AURKB (aurora kinase B), to regulate the central spindle assembly in endometrial cancers [30] . As expected, our functional enrichment results indicated that both of PRC1 and KIF23 were closely related to cell cycle associated processes such as mitotic cell cycle, nuclear division and M phase of mitotic cell cycle, which might support the hypothesis that PRC1 and KIF23 might exert significant roles in TAO via the cell cycle mediated regulation. What’s more, KIF23 was linked to PRC1 in the module, suggesting a potential regulatory relationship might exist between these two encoded genes.
MKI67 encodes a nuclear protein that is highly associated with cellular proliferation. Furthermore, this gene has even been used as a cell cycle marker for the prognosis of various diseases including Alzheimer disease [31] and hepatocellular carcinoma [32] .Interestingly, MKI67 is implied in the TAO disease via the interactions with chemicals such as peroxisome proliferator activated receptor-γ (PPAR-γ) and thiazolidinediones (TZDs),which were closely linked to TAO [33] . This provided a clue that MKI67 might be a potent biomarker of TAO detection.
Four crucial DEGs including SMC3 (structural maintenance of chromosomes 3) and ZNF263 were identified as TFs related to TAO. The gene SMC3 was also cell cycle related. The nuclear form of the SMC3 encoded protein is an essential element for the cohesion of sister chromatids during mitosis. Besides, the SMC3 acetylation is the basic mechanism in the regulation of sister chromatid cohesion [34] . Recruiting the method of logistic regression classifiers, SMC3 was observed implicated in the G2/M stage of the cell cycle [35] . In the present study, SMC3 was predicted as a TF correlated with TAO, implying that SMC3 might regulate other genes expression via the direct targeting.Unfortunately, rare evidence was existed to reveal the SMC3’s role as a TF, nor the corresponding targets. Therefore, our findings might provide a new insight into the role of SMC3 with regard to TAO. The ZNF263 (zinc finger protein 263) was generally considered to have an important role in basic cellular processes as a transcriptional repressor, but a recent study discovered that though embracing the KRAB domain which contributed to transcriptional repressor, the targets of ZNF263 had a high expression level, and the researchers proposed that ZNF263 might exert both positive and negative regulation of its targets as a TF [36] . Likewise, there was any studies reported the role of ZNF263 as a TF with regard to TAO but our results indicated that ZNF263 was a critical DEG and meanwhile was predicted as a TF related to TAO, implying it might have important roles in TAO events through the transcriptional regulation of its targets.
However, these are some limitations in our study. For examples,there are no enough samples from patients and have not been collected at present. In addition, there are also no suitable cell lines at present. Although all of the predicted results should be confirmed by laboratory data, the findings might provide a scientific guidance for future study and improve a further understanding of the progression of TAO.
In conclusion, our results identified several crucial genes related to TAO including TPX2 , CDCA5 , PRC1 and KIF23 ,which all might involve in cell cycle related process. Furthermore,regulatory relationships between TPX2 and CDCA5 as well as between PRC1 and KIF23 might exist. Additionally, MKI67 might be a potent biomarker of TAO, and SMC3 and ZNF263 might exert their roles as TFs in TAO progression.
ACKNOWLEDGEMENTS
Authors’ contributions: Conception and design of the research: Yang HB; Acquisition of data: Jiang J; Analysis and interpretation of data: Li LL; Statistical analysis: Yang HQ and Zhang XY; Drafting the manuscript: Yang HB; Revision of manuscript for important intellectual content: Yang HB and Jiang J.
Conflicts of Interest: Yang HB, None; Jiang J, None; Li LL, None; Yang HQ, None; Zhang XY, None.
REFERENCES
1 Maheshwari R, Weis E. Thyroid associated orbitopathy. Indian J Ophthalmol 2012;60(2):87-93.
2 Gopinath B, Wescombe L, Nguyen B, Wall JR. Can autoimmunity against calsequestrin explain the eye and eyelid muscle inflammation of thyroid eye disease? Orbit 2009;28(4):256-261.
3 Gould DJ, Roth FS, Soparkar CN. The diagnosis and treatment of thyroid- associated ophthalmopathy. Aesthetic Plast Surg 2012;36(3):638-648.
4 Bahn RS. Clinical review 157: pathophysiology of Graves’ ophthalmopathy:the cycle of disease. J Clin Endocrinol Metab 2003;88(5):1939-1946.
5 Boschi A. Quantification of cells expressing the thyrotropin receptor in extraocular muscles in thyroid associated orbitopathy. Br J Ophthalmol 2005;89(6):724-729.
6 Lehmann GM, Feldon SE, Smith TJ, Phipps RP. Immune mechanisms in thyroid eye disease. Thyroid 2008;18(9):959-965.
7 Tani J, Wall JR. Autoimmunity against eye-muscle antigens may explain thyroid-associated ophthalmopathy. CMAJ 2006;175(3):239.
8 Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, Leahy P, Li J, Andrade FH. Extraocular muscle is defined by a fundamentally distinct gene expression profile. Proc Natl Acad Sci U S A 2001;98(21):12062-12067.
9 Lahooti H, Parmar KR, Wall JR. Pathogenesis of thyroid-associated ophthalmopathy: does autoimmunity against calsequestrin and collagen XIII play a role? Clin Ophthalmol 2010;4(1):417-425.
10 Jublanc C, Beaudeux JL, Aubart F, Raphael M, Chadarevian R,Chapman MJ, Bonnefont-Rousselot D, Bruckert E. Serum levels of adhesion molecules ICAM-1 and VCAM-1 and tissue inhibitor of metalloproteinases, TIMP-1, are elevated in patients with autoimmune thyroid disorders: relevance to vascular inflammation. Nutr Metab Cardiovasc Dis 2011;21(10):817-822.
11 Płoski R, Szymański K, Bednarczuk T. The genetic basis of Graves’disease. Curr Genomics 2011;12(8):542-563.
12 Wescombe L, Lahooti H, Gopinath B, Wall JR. The cardiac calsequestrin gene (CASQ2) is up-regulated in the thyroid in patients with Graves’ ophthalmopathy-support for a role of autoimmunity against calsequestrin as the triggering event. Clin Endocrinol 2010;73(4):522-528.
13 Bolstad B. Preprocesscore: A collection of pre-processing functions. R package, version 1.20. 0. 2010
14 Smyth GK. Limma: linear models for microarray data. Bioinformatics and computational biology solutions using Rand bioconductor .Germany:Springer; 2005:397-420.
15 Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC,Lempicki RA. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol 2003;4(5):P3.
16 Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2011;39(Database issue):D561-D568.
17 Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 2010;27(3):431-432.
18 Nepusz T, Yu H, Paccanaro A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat Methods 2012;9(5):471-472.
19 Maere S, Heymans K, Kuiper M. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005;21(16):3448-3449.
20 Karolchik D, Barber GP, Casper J, et al . The ucsc genome browser database: 2014 update. Nucleic Acids Res 2014;42(Database issue):D764-D770.
21 Davis AP, Murphy CG, Johnson R, Lay JM, Lennon-Hopkins K,Saraceni-Richards C, Sciaky D, King BL, Rosenstein MC, Wiegers TC,Mattingly CJ. The comparative toxicogenomics database: update 2013. Nucleic Acids Res 2013;41(Database issue):D1104-D1114.
22 Brunet S, Dumont J, Lee KW, Kinoshita K, Hikal P, Gruss OJ, Maro B,Verlhac MH. Meiotic regulation of TPX2 protein levels governs cell cycle progression in mouse oocytes. PLoS One 2008;3(10):e3338.
23 Gruss OJ, Vernos I. The mechanism of spindle assembly functions of Ran and its target TPX2. J Cell Biol 2004;166(7):949-955.
24 Ma N, Titus J, Gable A, Ross JL, Wadsworth P. TPX2 regulates the localization and activity of Eg5 in the mammalian mitotic spindle. J Cell Biol 2011;195(1):87-98.
25 Mercier I, Casimiro MC, Wang C, et al . Human breast cancerassociated fibroblasts (CAFs) show caveolin-1 down-regulation and RB tumor suppressor functional inactivation: Implications for the response to hormonal therapy. Cancer Biol Ther 2008;7(8):1212-1225.
26 Kittler R, Pelletier L, Heninger AK, et al . Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat Cell Biol 2007;9(12):1401-1412.
27 Shrestha S, Wilmeth LJ, Eyer J, Shuster CB. PRC1 controls spindle polarization and recruitment of cytokinetic factors during monopolar cytokinesis. Mol Biol Cell 2012;23(7):1196-1207.
28 Subramanian R, Ti SC, Tan L, Darst SA, Kapoor TM. Marking and measuring single microtubules by PRC1 and kinesin-4. Cell 2013;154(2):377-390.
29 Neef R, Klein UR, Kopajtich R, Barr FA. Cooperation between mitotic kinesins controls the late stages of cytokinesis. Curr Biol 2006;16(3):301-307.
30 Chou WC, Cheng AL, Brotto M, Chuang CY. Visual gene-network analysis reveals the cancer gene co-expression in human endometrial cancer. BMC Genomics 2014;15:300.
31 Bonda DJ, Lee HP, Kudo W, Zhu X, Smith MA, Lee HG. Pathological implications of cell cycle re-entry in Alzheimer disease. Expert Rev Mol Med 2010;12:e19.
32 Hou YY, Cao WW, Li L, Li SP, Liu T, Wan HY, Liu M, Li X, Tang H. MicroRNA-519d targets MKi67 and suppresses cell growth in the hepatocellular carcinoma cell line QGY-7703. Cancer Lett 2011;307(2):182-190.
33 Lee S, Tsirbas A, Goldberg RA, McCann JD. Thiazolidinedione induced thyroid associated orbitopathy. BMC Ophthalmol 2007;7(1):8.
34 Zhang J, Shi X, Li Y, et al . Acetylation of SMC3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell 2008;31(1):143-151.
35 Ernst J, Plasterer HL, Simon I, Bar-Joseph Z. Integrating multiple evidence sources to predict transcription factor binding in the human genome. Genome Res 2010;20(4):526-536.
36 Frietze S, Lan X, Jin VX, Farnham PJ. Genomic targets of the KRAB and SCAN domain-containing zinc finger protein 263. J Biol Chem 2010;285(2):1393-1403.
Citation: Yang HB, Jiang J, Li LL, Yang HQ, Zhang XY. Biomarker identification of thyroid associated ophthalmopathy using microarray data. Int J Ophthalmol 2018;11(9):1482-1488
Correspondence to: Xiao-Yu Zhang. Department of Neurosurgery, the Second Affiliated Hospital of Harbin Medical University, No. 246 Xuefu Road, Nangang District, Harbin 150080, Heilongjiang Province, China. YuzhouZ88@163.com Received: 2017-09-05 Accepted: 2018-01-03
DOl: 10.18240/ijo.2018.09.09
Abstract · AlM: To uncover the underlying pathogenesis of thyroid associated ophthalmopathy (TAO) and explore potential biomarkers of this disease.· METHODS: The expression profile GSE9340, which was downloaded from Gene Expression Omnibus database,included 18 specimens from 10 TAO patients and 8 hyperthyroidism patients without ophthalmopathy. The platform was HumanRef-8 v2 Expression BeadChip. Raw data were normalized using preprocess. Core package and the differentially expressed genes (DEGs) were identified based on t-test with limma package of R. Functional enrichment analyses were performed recruiting the DAVlD tool. Based on STRlNG database, a protein-protein interaction (PPl) network was constructed, from which a module was extracted. The functional enrichment for genes in the module was performed by the BinGO plugin.· RESULTS: ln total, 861 DEGs (433 up-regulated and 428 down-regulated) between TAO patients and hyperthyroidism patients without ophthalmopathy were identified. Crucial nodes in the PPl network included TPX2, CDCA5, PRC1,KlF23 and MKl67, which were also remarkable in the module and all enriched in cell cycle process. Additionally,MKl67 was highly correlated with TAO. Besides, the DEGs of GTF2F1, SMC3, USF1 and ZNF263 were predicted as transcription factors (TFs).· CONCLUSlON: Several crucial genes are identified such as TPX2, CDCA5, PRC1 and KIF23, which all might play significant roles in TAO via the regulation of cell cycle process. Regulatory relationships between TPX2 and CDCA5 as well as between PRC1 and KIF23 may exist.Additionally, MKl67 may be a potent biomarker of TAO,and SMC3 and ZNF263 may exert their roles as TFs in TAO progression.