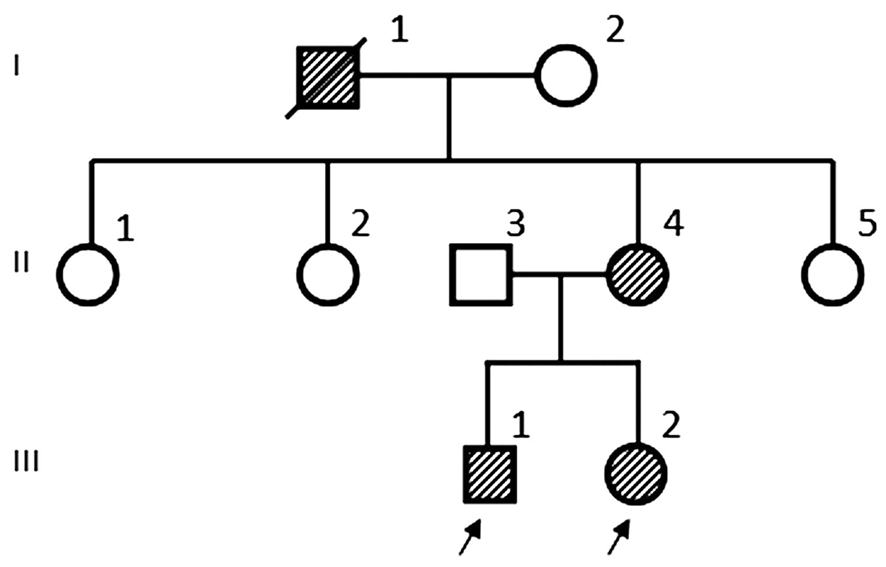
Figure 1 Pedigree of the Chinese NF1 family. III-1 and III-2 were the proband.
Jun Chen 1 , Bo Guo 1 , Min Ren 2 , Hong Lin 1 , Xin Zhang 1 , Si-Yi Chen 1 , Xiao-Tian Yu 1 , Zhu-Ping Xu 1
1 Department of Ophthalmology, West China Hospital, Sichuan University, Chengdu 610041, Sichuan Province, China
2 Department of Oncology, West China Hospital, Sichuan University, Chengdu 610041, Sichuan Province, China
Abstract · We analyzed the clinical features and NF1 gene mutation in a Chinese pedigree of neurofibromatosis type 1(NF1). Three members of this family were NF1 patients presenting with different clinical phenotypes and the others were asymptomatic. Exons of NF1 were amplified by polymerase chain reaction, sequenced, compared with a reference database. One novel NF1 frame-shift mutation c.703_704delTA, which resulted in a premature stop signal at codon 720 and the synthesis of truncated, was revealed.This mutation segregated with the NF1 members is likely responsible for the pathogenesis of NF1 in the family.
· KEYWORDS: neurofibromatosis type 1; NF1 gene; frameshift mutation
N eurofibromatosis type 1 (NF1) is one of the most common autosomal dominant disorders with equal sex incidence [1] . Inherited and sporadic cases occur in the population at a rate of about 1:3500, and about half of NF1 individuals have positive family history [2-3] . NF1 is characterized by diffuse café-au-lait macules, axillary freckling, neurofibromas,and iris Lisch nodules. NF1 is resulted from mutations in the NF1 gene, which has one of the highest rates of spontaneous mutations in the entire human genome [4] . So far, more than 1400 different mutations have been reported in the Human Gene Mutation Database (http://www.hgmd.org), and the point mutations are responsible for approximately 90% of cases of NF1 [5] . But no true “hot spots” have been found in NF1 [5] . NF1 gene encodes a large peptide with 2818 amino acids, called neurofibromin, which served as a regulator of signals for cell proliferation and survival, and in the absence of neurofibromin it may result in disturbances of organism development and uncontrolled cell proliferation [6] . Mutational analysis is therefore recommended to confirm the pathogenic mutation and for prenatal or pre-implantation genetic diagnosis.
In this study, a novel NF1 frame-shift mutation c.703_704delTA in exon 7 was revealed in this Chinese pedigree. This mutation was segregated with the disorder within the family and appeared to be the pathogenic mutation, which expands the spectrum of known NF1 mutations.
A three-generation Chinese pedigree (Figure 1) of eight individuals was recruited from West China Hospital of Sichuan University, China. We collected the clinical data and peripheral blood samples from seven family members, including three patients with NF1 (III:1, III:2, II:4) and four asymptomic members (I:2, II:2, II:3, II:5). We obtained the written informed consent from all the participating family members or their parents, and carried out this study according to the Declaration of Helsinki. This study was also approved by the Medical Ethics Committee of West China Hospital, Sichuan University.The seven members of the family underwent complete ophthalmologic and physical examinations. Diagnosis of NF1 was according to criteria created by National Institute of Health, while III-2 was diagnosed with NF1 by excisional biopsy.
Peripheral blood samples of these family members were collected, and then were stored at -85℃ in an ultra-low temperature freezer.A Qiamp Blood Kit (Qiagen, Hilden,Germany) was used to extract genomic DNA from 0.2 mL blood sample as described in the manufacturer’s instructions.One percent agarose gel electrophoresis was used to assess DNA integrity. NF1 exons were amplified from every participant’s genomic DNA by polymerase chain reaction(PCR) with forward and reverse primers. About 30 ng DNA,1× PCR buffer, 2.5 μmol/L MgCl 2 , 0.3 mmol/L of each of dNTPs, 1.5 U Pfu DNA polymerase, and 1.0 μmol/leach of the forward and reverse primers were contained in 30 μL PCR reaction mixture. The reagents were purchased from TaKaRa(Dalian, China). Reactions incubation was done at 94℃ for 2min before 35 cycles of denaturation at 94℃ for 20s, 50℃for 30s, and 72℃ for 2min, and an extension at 72℃ for 5min.The amplification products were purified with a cycle-pure kit (OMEGA; Bio-Tek, Doraville, GA, USA) and sequenced with an ABI 377XL automated DNA sequencer (Applied Biosystems, Foster City, CA, USA). The sequence data were pair-wise compared with the NF1 sequences in published literature. The neurofibromin protein sequences were aligned with a ClustalW tool (http://clustalw.ddbj.nig.ac.jp/top-e.html).
Figure 1 Pedigree of the Chinese NF1 family. III-1 and III-2 were the proband.
Clinical Findings The clinical manifestation of the patients were shown in Figure 2. The proband (III-1) was born with axillary freckling and scattered café-au-lait macules, which was the most marked on his neck. A soft subcutaneous mass, which substantially progressed in size, was touched under scalp since he was 4 years old. MR imaging revealed an extensive mass under the scalp, with hypointense on T1-weighted images,iso- to hyper-intense on T2-weighted images, and some cystic structure within the entity. Moreover, this patient was obviously shorter than normal. However, no tumor of central nerve system, dyskinesia or sensory disturbance was found.
The proband (III-2) was born with a subcutaneous mass in the left periorbital area. It was worth mentioning that a brown patch with localized hypertrichosis associated with NF was founded on the left forehead. Orbital CT revealed a huge subcutaneous soft tissue mass involving the left face, orbit and fossae termporalis, and typical sphenoid wing dysplasia.
II-4, the proband’s mother, was also a NF1 patient. Besides a large amount of café-au-lait macules and axillary freckling,lots of neurofibomas scattered on her whole body, with varying diameter from 3 mm to 5 cm. The proband’s grandfather (I-1)died over ten years ago. His manifestation was similar to that of II-4, with much little in size of subcutaneous neurofibromas.The asymptomatic family members, the proband’s father (II-3)and aunts (II-2 and II-5), appeared normal.
NF1 Gene Mutation Sequence analysis of NF1 revealed a novel frame-shift mutation c.703_704delTA, in exon 7 was founded in all patients, but not in the asymptomic family members (Figure 3). The c.703_704delTA NF1 mutation was cosegregated with the disease within this family. And no other mutation of NF1 was revealed.
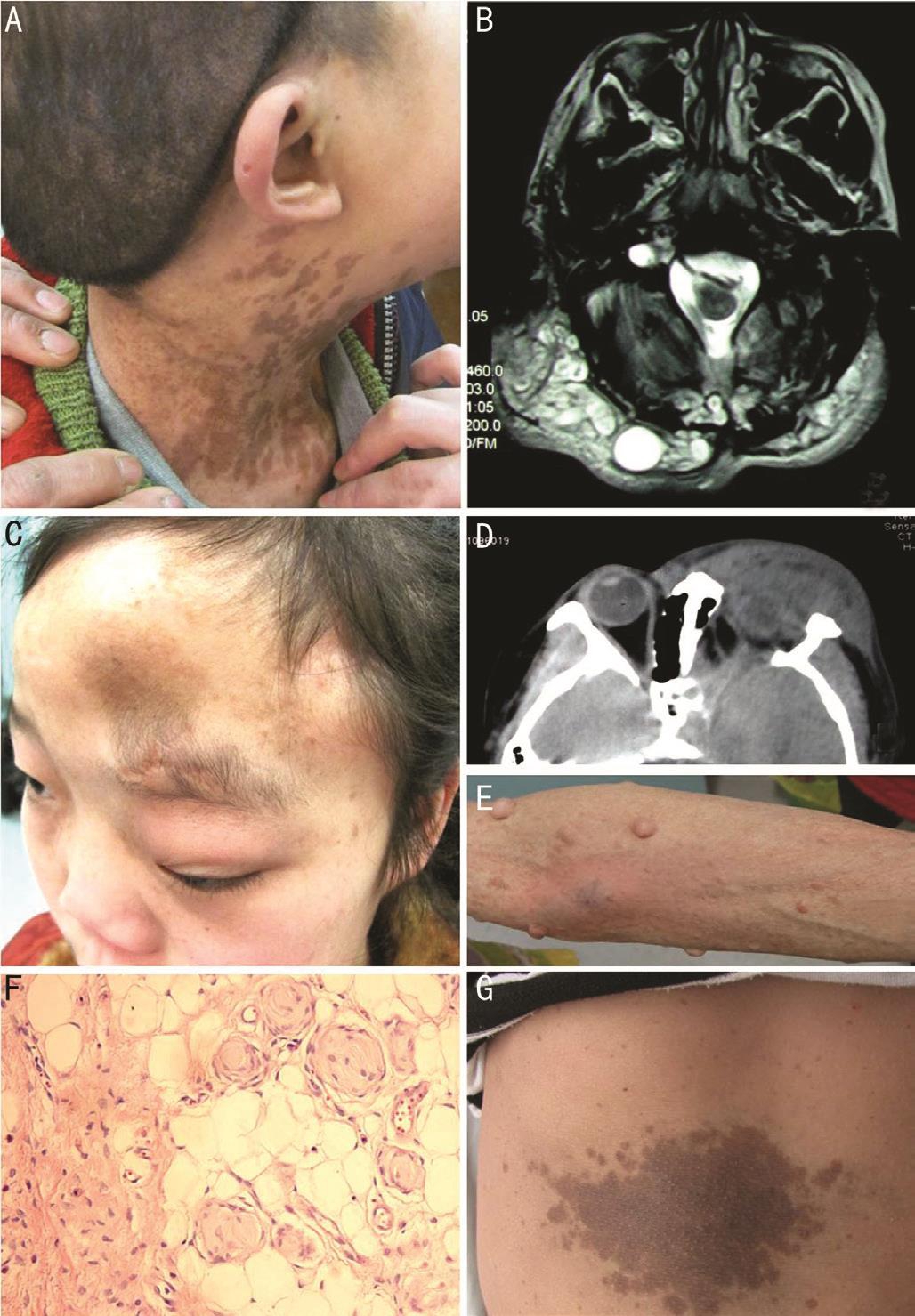
Figure 2 Clinical manifestations of patients in the Chinese family A: Photo of the proband (III-1) showed tumor under scalp and many café-au-lait macules on the neck; B: MR imaging of III-1 showed tumor was hyperintense on T2-weighted image; C: Photo of the proband (III-2) showed tumor involved orbit, face, forehead and fossae termporalis; D: Typical sphenoid wing dysplasia of III-2 on CT; E: Photo of II-4 showed subcutaneous neurofibromas and multiple café-au-lait macules scattered on her right arm; F: Biopsy of III-2 (HE staining,magnification 400×) revealed that the main entity of neurofibroma is composed of spindle and oval Schwann cells; G: Photo of the proband(III-2) showed big café-au-lait macule on her trunk.
Bioinformatic Analysis The deletion of a thymine and adenine in codon 703 and 704 of NF1 exon 7 caused frame shift and formed a termination codon (TGA) that introduced a premature stop signal at codon 720. This resulted in the termination of the amino acid sequence starting at position 234 of neurofibromin (Figure 4).
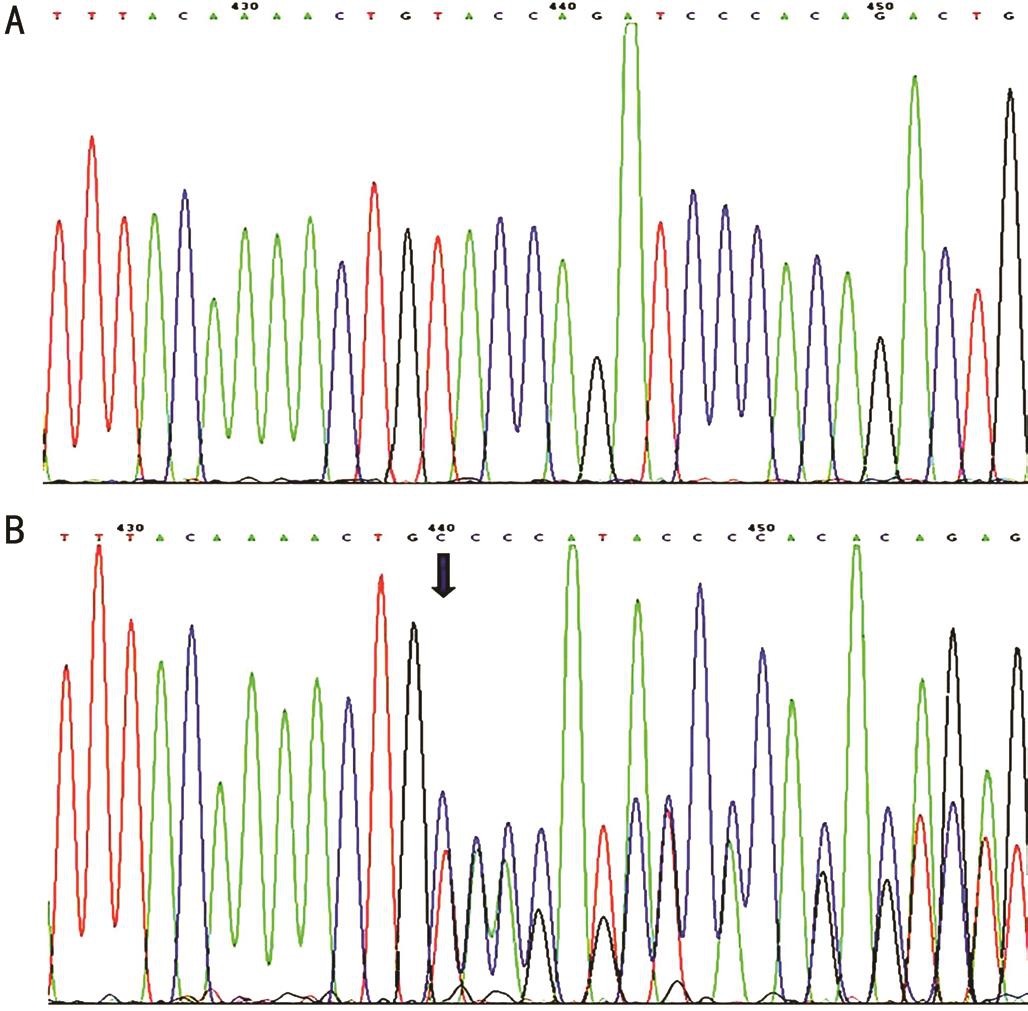
Figure 3 Sequencing results of the NF1 gene A: Wild-type sequence from an asymptomic member; B: Frame-shift mutation c.703_704delTA of the NF1 gene (arrow) found in all patients.
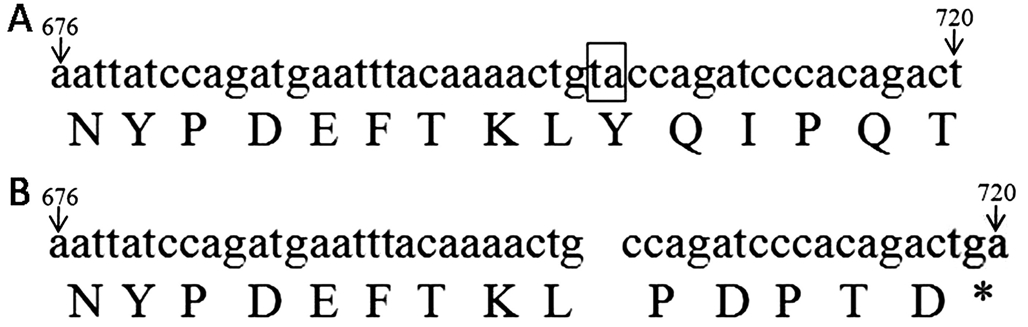
Figure 4 Base sequence of NF1 gene from 676 to 720 and its corresponding amino acid A: Base sequence of normal allele; B:Base sequence of mutation allele. The thymine and adenine in codon 703 and 704 was deleted and formed a termination codon in codon from 718 to 720 (thymine, guanine and adenine).
Our study revealed a novel frame-shift mutation c.703_704delTA in exon 7 of NF1 gene in a Chinese family with NF1. The mutation cosegregated with the NF1 phenotype in this family,and was not founded in the public databases and the unaffected members of the family, implying that this frame-shift mutation was disease-causing. Bioinformatic analysis indicates that the mutation caused a premature stop signal at codon 720 and the termination of the amino acid sequence at position 234 of neurofibromin, which also resulted in a truncated protein of 240-amino acid residues instead of full-length neurofibromin.In a recent study, Cai et al [7] showed an adjacent frame-shift mutation (c.702_703delGT) in exon 7 in a Chinese family with NF1, which was also resulted from the formation of abnormal neurofibromin with truncated 240 amino acid residues. Meanwhile, abundant previous studies have also indicated that most NF1 patients were caused by mutations of the NF1 gene that lead to the synthesis of truncated, nonfunctional neurofibromin protein. The impaired neurofibromin functioning will affect the normal cellular function and result in the occurrence of clinical signs of NF1 [6,8-9] . The basic function of neurofibromin protein is modulation of the RAS protein activity, which is necessary for regulating cell proliferation and differentiation. Neurofibromin inactivates the active RAS-GTP and its signal transduction pathways by the RAS-GTPase activating protein related domain (GRD),that can stimulate the intrinsic GTPase of p21-RAS-GTP to hydrolyze GTP to GDP [6,8,10] . Increased RAS-GTP leads to cell proliferation through Raf kinase and protects cells from apoptosis by activating protein kinase B/Akt [10] . In addition,neurofibromin can regulate the activity of adenylate cyclase,which may interfere with the cell cycle progression through the signal pathway of cAMP/protein kinase [11] . Neurofibromin also interacts with many other proteins that are engaged in morphogenesis of neural cells (syndecans, CRMP proteins),actin cytoskeleton rearrangements (LIMK2, Rho and Rac) and intracellular transport (tubulin, kinesin) [10] . With neurofibromin being a tumor suppressor, it explains why that patients with NF1 harbor an increased risk for developing both benign and malignant tumors [12] . About 50% of NF1 patients begin to show clinical symptoms within the first year, 97% of patients become apparent before they reach 8 years old, and the penetrance is virtually 100% by the age of 20y [6,13] . However,researchers have found little evidence suggesting genotypephenotype correlation in NF1 [14-15] . Our study also reflects this uncorresponding genotype-phenotype relation. By now, two correlations between the clinical phenotype and the mutant alleles in the NF1 gene have been observed that somatic mosaicism in NF1 was thought to be the cause of segmental neurofibromatosis [16] , and NF1 micro-deletion usually presents more severe clinical phenotypes, including a higher prevalence of learning disabilities and dysmorphic features [17] . The marked phenotypic variation associated with NF1 even in individuals carrying the same NF1 mutation suggest that other factors,including feature-specific modifier genes, environmental factors and stochastic factors, are likely involved in determining clinical manifestations [18] .
In conclusion, the novel c.703_704delTA NF1 mutation appears to be the cause of NF1 in this pedigree. The mutation further proves that the loss or diminished function of neurofibromin may lead to NF1. A pathogenic mutation in the NF1 should be added to the list of diagnostic criteria, as mutational analysis could provide precise genetic counseling.Much additional research is needed to determine the related genes and factors regulating the phenotype of NF1 and to understand their mechanisms.
ACKNOWLEDGEMENTS
Foundation: Supported by the National Major Scientific Equipment Program (No.2012YQ12008005).
Conflicts of Interest: Chen J, None; Guo B, None; Ren M, None; Lin H, None; Zhang X, None; Chen SY, None; Yu XT, None; Xu ZP, None.
REFERENCES
1 Ferner RE, Gutmann DH. Neurofibromatosis type 1 (NF1): diagnosis and management. Handb Clin Neurol 2013;115:939-955.
2 Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol 2005;141(1):71-74.
3 North K. Neurofibromatosis type 1: review of the first 200 patients in an Australian clinic. J Child Neurol 1993;8(4):395-402.
4 Bikowska-Opalach B, Jackowska T. Neurofibromatosis type 1-description of clinical features and molecular mechanism of the disease. Med Wieku Rozwoj 2013;17(4):334-340.
5 Yap YS, McPherson JR, Ong CK, Rozen SG, Teh BT, Lee AS,Callen DF. The NF1 gene revisited-from bench to bedside. Oncotarget 2014;5(15):5873-5892.
6 Abramowicz A, Gos M. Neurofibromin in neurofibromatosis type 1-mutations in NF1 gene as a cause of disease. Dev Period Med 2014;18(3):297-306.
7 Cai SP, Fan N, Chen J, Xia ZL, Wang Y, Zhou XM, Yin Y, Wen TL, Xia QJ, Liu XY, Wang HY. A novel NF1 frame-shift mutation(c.702_703delGT) in a Chinese family with neurofibromatosis type 1. Genet Mol Res 2014;13(3):5395-5404.
8 Hirata Y, Brems H, Suzuki M, Kanamori M, Okada M, Morita R,Llano-Rivas I, Ose T, Messiaen L, Legius E, Yoshimura A. Interaction between a domain of the negative regulator of the Ras-ERK pathway,SPRED1 protein, and the GTPase-activating protein-related domain of neurofibromin is implicated in legius syndrome and neurofibromatosis type 1. J Biol Chem 2016;291(7):3124-3134.
9 Corsello G, Antona V, Serra G, Zara F, Giambrone C, Lagalla L,Piccione M, Piro E. Clinical and molecular characterization of 112 single-center patients with neurofibromatosis type 1. Ital J Pediatr 2018;44(1):45.
10 Abramowicz A, Gos M. Neurofibromin-protein structure and cellular functions in the context of neurofibromatosis type I pathogenesis. Postepy Hig Med Dosw 2015;69:1331-1348.
11 Brown JA, Diggs-Andrews KA, Gianino SM, Gutmann DH.Neurofibromatosis-1 heterozygosity impairs CNS neuronal morphology in a cAMP/PKA/ROCK-dependent manner. Mol Cell Neurosci 2012;49(1):13-22.
12 Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist 2012;17(1):101-116.
13 Scalzone M, Coccia P, Ruggiero A, Riccardi R. Neurofibromatosis type 1 clinical features and management. Pediatr Med Chir 2009;31(6):246-251.
14 Sabbagh A, Pasmant E, Imbard A, Luscan A, Soares M, Blanché H,Laurendeau I, Ferkal S, Vidaud M, Pinson S, Bellanné-Chantelot C,Vidaud D, Parfait B, Wolkenstein P. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: the French experience. Hum Mutat 2013;34(11):1510-1518.
15 Ejerskov C, Farholt S, Skovby F, Vestergaard EM, Haagerup A.Clinical presentations of 23 half-siblings from a mosaic neurofibromatosis type 1 sperm donor. Clin Genet 2016;89(3):346-350.
16 Castle B, Baser ME, Huson SM, Cooper DN, Upadhyaya M.Evaluation of genotype-phenotype correlations in neurofibromatosis type 1. J Med Genet 2003;40(10):e109.
17 Zhu L, Zhang Y, Tong H, Shao M, Gu Y, Du X, Wang P, Shi L, Zhang L,Bi M, Wang X, Zhang G. Clinical and molecular characterization of NF1 patients: single-center experience of 32 patients from China. Medicine(Baltimore) 2016;95(10):e3043.
18 Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol 2000;151(1):33-40.
Citation: Chen J, Guo B, Ren M, Lin H, Zhang X, Chen SY, Yu XT, Xu ZP. A novel NF1 frame-shift mutation c.703_704delTA in a Chinese pedigree with neurofibromatosis type 1. Int J Ophthalmol 2018;11(9):1562-1565
Received: 2017-03-19 Accepted: 2018-06-28
DOl: 10.18240/ijo.2018.09.22
Correspondence to: Zhu-Ping Xu. No.37, Guoxue Alley,Wuhou District, Chengdu 610041, Sichuan Province, China.xuzp@hotmail.com