
・Basic Research・
Mutation analysis of FBN1 gene in two Chinese
families with congenital ectopia lentis in northern China
Su-Zhen Tang1, Ya-Ning
Liu1, Shao-Hua Hu1, Hao Chen1, Hui Zhao2,
Xue-Mei Feng1, Xiao-Jing Pan3, Peng Chen1
1Department of
Human Anatomy, Histology and Embryology, School of Basic Medicine, Qingdao
University, Qingdao 266071, Shandong Province, China
2The 971
Hospital of the Chinese People’s Liberation Army Navy, Qingdao 266071, Shandong
Province, China
3Qingdao Eye
Hospital, Shandong Eye Institute, Shandong First Medical University &
Shandong Academy of Medical Sciences, Qingdao 266071, Shandong Province, China
Co-first authors: Su-Zhen Tang and Ya-Ning Liu
Correspondence to: Peng Chen. Department of Human Anatomy, Histology and Embryology, School of
Basic Medicine, Qingdao University, 308 Ningxia Road, Qingdao 266071, Shandong
Province, China. chenpeng599205@126.com; Xiao-Jing Pan. Qingdao Eye Hospital,
Shandong Eye Institute, Shandong First Medical University & Shandong
Academy of Medical Sciences, 5 Yan’er dao Road, Qingdao 266071, Shandong
Province, China. panxjcrystal@163.com
Received:
Abstract
AIM: To summarize the phenotypes
and identify the underlying genetic cause of the fibrillin-1 (FBN1)
gene responsible for congenital ectopia lentis (EL) in two Chinese families in
northern China.
METHODS: A detailed family history and
clinical data from all participants were collected by clinical examination. The
candidate genes were captured and sequenced by targeted next-generation
sequencing, and the results were confirmed by Sanger sequencing. Haplotyping
was used to confirm the mutation sequence. Real-time PCR was used to determine
the FBN1 messenger ribonucleic acid (mRNA) levels in patients with EL
and in unaffected family members.
RESULTS: The probands and other patients
in the two families were affected with congenital isolated EL. A heterozygous FBN1 mutation in exon 21 (c.2420_IVS20-8 delTCTGAAACAinsCGAAAG) was identified
in FAMILY-1. A heterozygous FBN1 mutation in exon 14 (c
CONCLUSION: The insertion-deletion mutation
(c.2420 IVS20-8delTCTGAAACA insCGAAAG) in the FBN1 gene is first identified in isolated EL. The mutation (c
KEYWORDS: congenital ectopia lentis; autosomal
dominant; targeted next-generation sequencing; FBN1; fibrillin-1
DOI:10.18240/ijo.2019.11.02
Citation: Tang SZ, Liu YN, Hu SH, Chen H, Zhao H, Feng XM, Pan XJ,
Chen P. Mutation analysis of FBN1 gene in two Chinese families
with congenital ectopia lentis in northern China. Int J Ophthalmol
2019;12(11):1674-1679
INTRODUCTION
Ectopia lentis (EL; OMIM 129600) is characterized by a displacement or
malposition of the optic lens from its normal location and the zonular
filaments are stretched or discontinued[1]. Most EL cases are associated with Marfan syndrome
(MFS; OMIM 154700), an autosomal dominant disease that includes cardiovascular,
skeletal, and ocular system abnormalities[2].
The clinical manifestations of isolated EL are mild or severe. The main
symptoms include refractive error, amblyopia, complex glaucoma or retinal
detachment. EL seriously affects visual quality. It is the second most frequent
cause of lens surgery in juveniles[3].
FBN1 is located
on chromosome 15q21.1. Mutations in FBN1 can cause isolated or
predominant EL[4].
Fibrillin 1 is a cysteine-rich glycoprotein that is broadly distributed in
elastic and nonelastic connective tissues[5-6].
Both syndromic and isolated EL have strong genetic heterogeneity.
Pathogenic variants in FBN1[7] can cause connective tissue disorders such as MFS and
autosomal dominant EL. To date, the Universal Mutation Database (UMD)-FBN1
database (http://www.umd.be/FBN1/) have registered over 600 FBN1 mutations[8]. It is vital to
isolate EL and its related diseases by genotype and phenotype correlations. The
study of molecular genetics of FBN1 contributes to the development of
prenatal diagnosis of this gene-related disease, and also contributes to the
early diagnosis and risk prediction of high-risk patients.
Isolated EL pedigree has been reported many times in different races[7-10]. Isolated EL may be an
independent subtype caused by specific FBN1 mutations or other
regulatory factors. We recruited two Chinese pedigrees affected with isolated
EL. Mutation in the FBN1 gene (c
SUBJECTS AND METHODS
Ethical Approval The study was conducted in
accordance with the principles of the Declaration of Helsinki. Informed consent
was obtained from all the participants.
Clinical Examination The two autosomal dominant EL
families came from Qingdao (Shandong Province, China). All family members
included in the study had received comprehensive medical history review,
ophthalmic examination. Two hundred individuals in the control group were
healthy.
FAMILY-1 (four generations) had sixteen individuals (seven affected and
nine unaffected, ten males and six females). There were thirty-two individuals
in FAMILY-2 (five generations, ten affected and twenty-two unaffected, nineteen
males and thirteen females). FAMILY-1 and FAMILY-2 family members do not have
diseases of other systems other than the visual system.
Targeted Next-generation Sequencing
Whole blood
genomic DNA extraction was performed with DNA extraction kit (Tiangen, Beijing,
China) from venous blood. Inheritable genetic vision system-related genes were
captured as described[10].
The probands (IV:
Variant Analysis and Verification
According to
the reference genome, data were analyzed and provided as described[10]. After variant
annotation, we primarily analyzed the nonsynonymous variants, coding indels,
splice site variants. Exome data were filtered by the public databases (1000
Genomes Project, dbSNP, YH database, and HapMap 8 database).
Sanger sequencing was used to sequence the mutation sites selected by the
filtration. Haplotyping was used to confirm the mutation sequence as described[10].
Ribonucleic Acid Extraction and
Real-time Polymerase Chain Reaction
Real-time PCR was performed using
SYBR Premix Ex Taq kit (Tiangen). FBN1 primer sequences were
RESULTS
Clinical Findings The two families in this study lived
in northern China. FAMILY-1 was an autosomal dominant four-generation family
with a total of 16 members, of which 6 were affected by bilateral congenital EL
(Figure
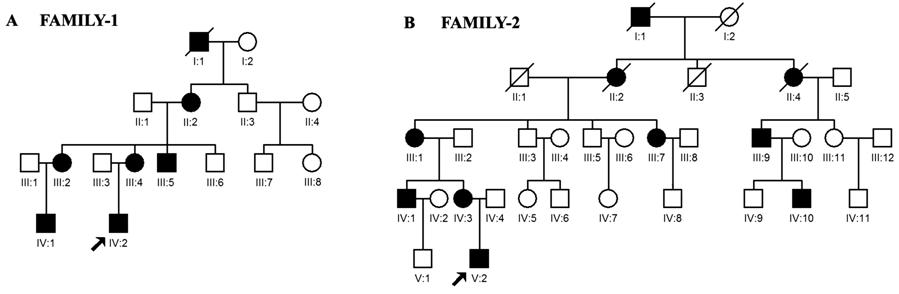
Figure 1 Pedigrees of the two
Chinese families with autosomal dominant congenital ectopia lentis Squares
indicates males, and circles indicates females. The affected members are
represented by filled symbols. Slashes (/) indicate the deceased individuals.
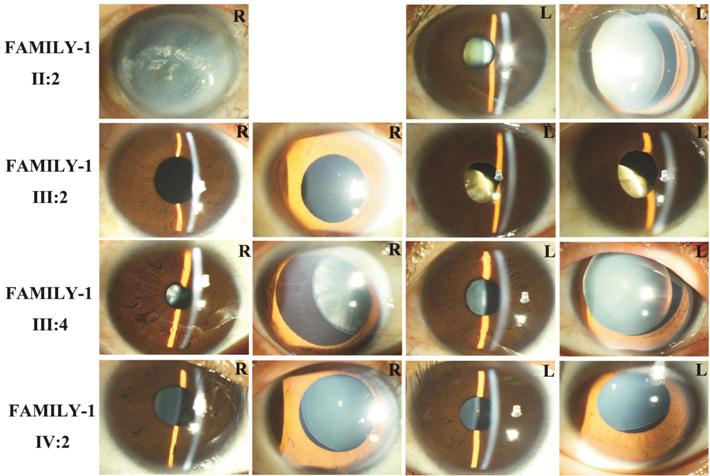
Figure 2 Photographs of patients in FAMILY-1 R: Right eye; L: Left eye.
We identified another five-generation family with ten confirmed individuals
affected with autosomal dominant EL (Figure 1B). Bilateral nasal dislocations
were detected in the seven living patients (Figure 3). The onset age of
patients with EL was around 6 to 17y.
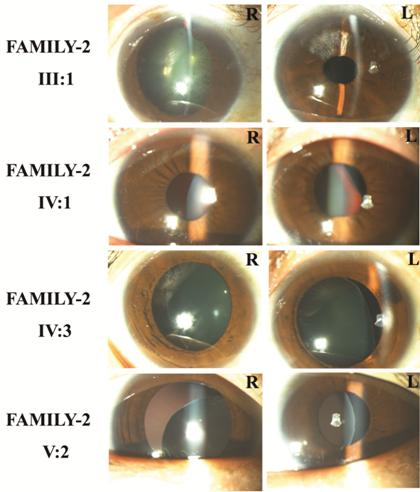
Figure 3 Photographs of the patients in FAMILY-2 R: Right eye; L: Left eye.
Associated Gene FBN
Verification of Candidate Gene FBN1 by Sanger Sequencing Only a heterozygous mutation
(c.2420-IVS20-8delTCTGAAACAinsCGAAAG) was detected in exon 21 of the six
affected individuals in FAMILY-1 (Figure 4) by using Sanger sequencing. Only a
heterozygous mutation (c

Figure 4 Sequence chromatograms of the detected fibrillin 1 mutations in
FAMILY
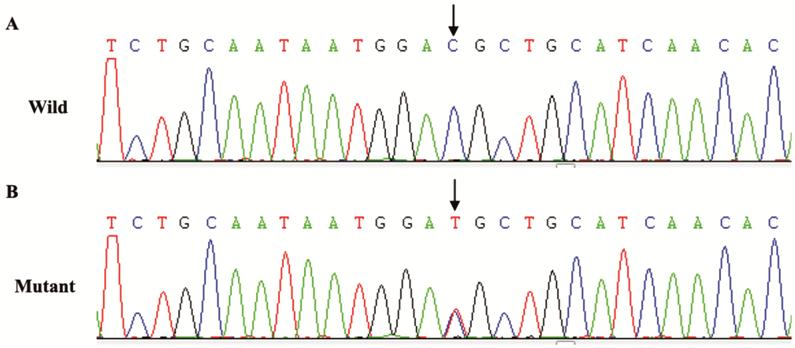
Figure 5 Sequence chromatograms
of the detected fibrillin 1 mutations in FAMILY
c.2420-IVS20-8delTCTGAAACAinsCGAAAG
and c
FBN1 Gene Expression in Patients with EL in FAMILY-1 FBN1 mRNA
expression was detected in EL patients and normal members in FAMILY-1. The
level of FBN1 mRNA in EL patients is 52% of that of unaffected members
in the family (P=0.01).
DISCUSSION
It is suggested that lens
ectopicity may not be an independent diagnosis, but may be a mild manifestation
of a broad clinical symptom spectrum of MFS[12-14]. In some cases, ectopic lens may be one of the signs
of some syndromes, so metabolic screening and DNA testing have developed into
an effective diagnostic method for distinguishing isolated EL from syndromes[15-16]. By using next generation
sequencing (NGS), multiple genes can be analyzed simultaneously and with high
precision. The cost of this targeted approach has been greatly reduced, and the
advantages of rapid detection and analysis are currently being used in standard
clinical diagnostics. Differential diagnosis of EL and syndromic EL has
important clinical significance, including patient prognosis, monitoring and
prevention of potentially life-threatening complications. Moreover, genetic
diagnosis of EL is critical to determining the genetic pattern and risk of
recurrence of family members. In addition, a clear genetic diagnosis can help
patients to consider reproductive options, and help relatives to perform
pre-symptomatic DNA testing. This study was performed in two EL families in
northern China by using NGS.
A novel insertion deletion mutation (c.2420_IVS20-8 delTCTGAAACAinsCGAAAG,
a heterozygous mutation) in FBN1 gene in FAMILY-1 was reported in a
Chinese family in this study. The insertion site found in FAMILY-1 is located
in the cbEGF domain of FBN1 protein, leading to early termination of
translation and possibly affecting the binding of calcium to cbEGF. The
clinical significance of this mutation is currently unknown, and further
pedigree analysis and functional studies are needed to verify whether it is a
pathogenic mutation. However, according to previous reports, the mutation has a
high probability of pathogenicity. In addition, no similar nucleotide changes
were detected in normal individuals in the family and 200 normal Chinese
controls. And the mutation was filtered by the FBN1 SNP database. Many FBN1
mutations have been reported in the Chinese population[17], and the clinical phenotypes
caused by various FBN1 mutations are different. The missense mutation c
In a MFS family, people with this mutation have three different cardinal
phenotypes (aortic dissection, EL and unaffected)[19].
FBN1, located on chromosome15q21, encodes a fibrinogen
protein with a molecular weight of approximately 350-kDa. Fibrin-1 encoded by FBN1
is the major structural element in the lens suspensory ligament. Fibrin-1 is
involved in the formation of the lens suspensory ligament[20], which is mainly secreted by
non-pigment cells in the ciliary body.
FBN1 mutations can cause type 1 fibrillinopathies and MFS.
Type 1 fibrillinopathies include Marchesani syndrome (MASS), isolated EL,
isolated skeletal features of MFS, and thoracic aortic aneurysms[21]. To date, over
600 mutations in FBN1 have been reported. In addition to neonatal MFS,
no correlation has been identified between genotypes/phenotypes[22].
In addition, recent studies have
shown that cysteine substitutions, rather than the location of amino acids in
protein sequences, are closely related to isolated or predominant EL[23].
In conclusion, we found a novel insertion deletion mutation
(c.2420-IVS20-8delTCTGAAACA insCGAAAG, a heterozygous mutation) in FBN1 gene
in FAMILY-1 with congenital EL and a known point mutations (c
ACKNOWLEDGEMENTS
Foundations: Supported by Natural Science Foundation of Shandong Province
(No.ZR2018MH016); China Postdoctoral Science Foundation Funded Project (No
Conflicts of Interest: Tang SZ, None; Liu YN, None; Hu SH, None; Chen
H, None; Zhao H, None; Feng XM, None; Pan XJ, None; Chen
P, None.
REFERENCES