
Citation: Song N, Leng L, Yang XJ, Zhang YQ, Tang C, Chen WS, Zhu
W, Yang X. Compound heterozygous mutations in CYP1B1 gene
leads to severe primary congenital glaucoma phenotype. Int J Ophthalmol
2019;12(6):909-914
DOI:10.18240/ijo.2019.06.05
・Basic Research・
Compound
heterozygous mutations in CYP1B1 gene leads to severe primary congenital
glaucoma phenotype
Na Song1, Lin Leng1, Xue-Jiao Yang1,
Yu-Qing Zhang1, Chun Tang1, Wen-Shi Chen1, Wei
Zhu2, Xian Yang1
1Department
of Ophthalmology, the Affiliated Hospital of Qingdao University, Qingdao
266003, Shandong Province, China
2Department
of Pharmacology, School of Pharmacy, Qingdao University, Qingdao 266021,
Shandong Province, China
Correspondence to: Xian Yang. Department of Ophthalmology, the Affiliated
Hospital of Qingdao University, Qingdao 266003, Shandong Province, China.
yangxian_zhao@126.com; Wei Zhu. Department of Pharmacology, School of Pharmacy,
Qingdao University, Qingdao 266021, Shandong Province, China. wzhu@qdu.edu.cn
Received: 2018-09-20
Accepted: 2018-12-15
Abstract
AIM: To identify the novel mutation alleles in the CYP1B1 gene of
primary congenital glaucoma (PCG) patients at Shandong Province of China, and
investigate their correlation with glaucomatous features.
METHODS: The DNA from the peripheral blood of 13 congenital
glaucoma patients and 50 ethnically matched healthy controls from the
affiliated hospital of Qingdao University were extracted. The coding region of
the CYP1B1 gene was amplified by PCR and direct DNA sequencing was
performed. Disease causing-variants were analyzed by comparing the sequences
and the structures of wild type and mutant CYP1B1 proteins by PyMOL
software.
RESULTS: Two missense mutations, including A330F caused by c.988G>T&c.989C>T, and R390H caused by c.1169G>A, were identified in one of
the 13 PCG patients analyzed in our study. A330F mutation was observed to be novel in the Chinese
Han population, which dramatically altered the protein structure of CYP1B1
gene, including the changes in the ligand-binding pocket. Furthermore, R390H
mutation caused the changes in heme-protein binding site of this gene. In
addition, the clinical phenotype displayed by PCG patient with these mutations
was more pronounced than other PCG patients without these mutations. Multiple
surgeries and combined drug treatment were not effective in reducing the
elevated intraocular pressure in this patient.
CONCLUSION: A novel A330F
mutation is identified in the CYP1B1 gene of Chinese PCG patient.
Moreover, in combination with other mutation R390H, this PCG patient shows
significant difference in the CYP1B1 protein structure, which may
specifically contribute to severe glaucomatous phenotype.
KEYWORDS: primary congenital glaucoma; CYP1B1
gene; missense mutation; protein structure
DOI:10.18240/ijo.2019.06.05
Citation: Song N, Leng L, Yang XJ, Zhang YQ, Tang C, Chen WS, Zhu
W, Yang X. Compound heterozygous mutations in CYP1B1 gene
leads to severe primary congenital glaucoma phenotype. Int J Ophthalmol
2019;12(6):909-914
Outline
INTRODUCTION
SUBJECTS
AND METHODS
RESULTS
DISCUSSION
ACKNOWLEDGEMENTS
REFERENCES
INTRODUCTION
Primary
congenital glaucoma (PCG), alternatively known as developmental glaucoma, is a
developmental eye defect, primarily caused by the abnormal development of
anterior chamber angle during neonatal or infantile stage[1].
Typically, most patients display glaucomatous phenotypes during birth, but in
some cases its onset has also been observed at adolescence[2].
Typical symptoms of PCG include tearing, photophobia, and blepharospasm[3]. In absence of any treatment, ocular hypertension can
lead to irreversible damage to the optic nerve, thereby resulting in reduced
vision or subsequent complete blindness.
Congenital
glaucoma has been shown to be a hereditary disease transmitted in an autosomal
recessive pattern[4]. Although it’s complete
etiology has not been thoroughly elucidated, genetic cause is believed to be
the most important risk factor in PCG patients. Since the first genetic study
in PCG patients[5], multiple genetic loci in the
gene GLC3 have been identified, including GLC3A[5], GLC3B[6], GLC3C,
and GLC3D[7]. These studies have shown the
presence of two pathogenic genes, cytochrome P450 1B1 (CYP1B1)
and latent-transforming growth factor beta-binding protein 2 (LTBP2)
on these genetic loci. The mutation in the CYP1B1 gene is considered as
the most common pathological reason for congenital glaucoma. Typically, CYP1B1
protein structure display 4 conserved helix bundles, including J-helix,
K-helix, β-sheets, meander region, and heme-binding region[8].
In addition, the N-terminus hinge region and C-terminus conserved core
structures (CCSs) have been reported to be the most crucial regions for
maintaining the fundamental properties of CYP1B1[2,9]. Interestingly, the clinical reports from different
ethnic groups showed that PCG patients, with CYP1B1 mutations, like
missense mutations of W57C[9] and G365W[10] in the
N-terminus hinge region or CCSs, generally have more severe glaucomatous
phenotypes. To date, more than 150 mutation variants of CYP1B1 gene have
been found in congenital glaucoma patients worldwide[11].
In Chinese patients, 43 mutation variants have been reported, including R390H,
the common mutation identified in PCG patients from all ethnic groups, and
L107V mutations uniquely identified in Chinese PCG patients only[12].
Based on
this information, we in our study have tried to investigate if any novel CYP1B1
gene mutations exist in PCG patients from Shandong province of China. To
address it, we analyzed the CYP1B1 gene individually from 13 PCG
diagnosed Chinese patients, and 50 healthy controls from Shandong Province by
direct sequencing of its coding region. The structure of the mutated protein
was analyzed through PyMOL software (USA). The severe glaucomatous phenotype of
one PCG patient with novel mutation indicated the important role played by
specific mutational allele in regulating CYP1B1 gene function during
development.
SUBJECTS AND METHODS
Ethical
Approval The study was conducted in
accordance with the Declaration of Helsinki and was approved by the Ethics
Committee of the Affiliated Hospital of Qingdao University. The informed
consents were obtained from each subject or his/her guardian for their
participation in this study.
The ocular
examination of the sporadic PCG patients and ethnically matched healthy
controls were performed at Affiliated Hospital of Qingdao University. The
examination included vision tests, intraocular pressure (IOP), slit lamp
bio-microscopy, corneal diameter, and cup-to-disc ratio. Based on these
clinical reports, patients with other ocular diseases beside PCG or systemic
illnesses were excluded from this study. Based on the IOP, 13 sporadic PCG
patients with IOP of higher than 21 mm
Hg, and 50 healthy controls with IOP lower than 21 mm Hg, were eventually selected for this study.
Peripheral
blood samples were collected from all subjects. Leukocyte DNA were extracted
using Massive Whole Blood Genomic DNA Extraction Kit (BioTeke, Beijing, China).
The coding region of CYP1B1 gene was amplified from the genomic DNA by
polymerase chain reaction (PCR). The PCR reaction was performed in a 25 µL
reaction mixture containing 0.1 µg genomic DNA, 40 µmol/L forward and reverse
primers (Table 1), 3 mmol/L magnesium chloride and 2×HieffTM PCR
Master Mix (YESEN Biotech Co., Shanghai, China). The following PCR conditions were
used for amplification: an initial denaturation step at 95℃ for 5min, 32 cycles of denaturation
at 95℃
for 30s, annealing temperature for 30s, and extension at 72℃ for 30s, with final additional
extension time of 10min at 72℃.
Table 1 The
primers to amplify coding regions of CYP1B1 gene
|
Name
|
Sequence (5’
to 3’)
|
Size of the products (bp)
|
|
CYP1B1-2F
|
TCAGCTCCGACCTCTCCACCCA
|
1316
|
|
CYP1B1-2R
|
AGTCCCTTTACCGACGCGATCT
|
|
|
CYP1B1-3F
|
TTCTTA AAGTCCATCTTGTAAT
|
997
|
|
CYP1B1-3R
|
AAAAAAATCTCCCAGAAGCTCC
|
|
CYP1B1: Cytochrome P450 Family 1 Subfamily
B Member 1.
The
amplified PCR products were subjected to agarose gel electrophoresis and the
target PCR fragments were extracted from Biowest Agarose by QIAquick Gel
Extraction Kit (QIAGEN, Shanghai, China). Next, the direct sequencing of the
amplified product was completed using amplification primers (Ruibo Xingke
Biotech Co. Beijing, China), and the sequences were analyzed using Chromas
V2.3, DNAman V7.0 software along with PubMed Blast
(https://blast.ncbi.nlm.nih.gov/Blast.cgi) search engine.
In addition,
the sequence of wild-type CYP1B1 gene was downloaded from
https://www.rcsb.org, and saved as PDB file. The mutated amino acids in the CYP1B1
protein were changed manually using ICMpro software, and the PDB files were
uploaded into PyMOL software to generate 3D structure of the protein. The
mutated amino acids and key ligands were labeled in different colors.
RESULTS
Among the total
13 primary congenital glaucoma patients included in our study, 12 were males
and 1 female, with age ranging from 3 to 23y. The median age at the time of
diagnosis was 3mo. In addition, one patient had unilateral ocular abnormality
diagnosis, while all other 12 patients were diagnosed with bilateral congenital
glaucoma. The average IOP prior to surgeries in these patients was 37.19 mm Hg and the mean corneal
diameter was 13 mm.
The analysis
of CYP1B1 gene led to the identification of two missense mutations in
one of the 13 PCG patients through direct sequence analysis, including c.988G>T [G=0.00000 (0/112670, ExAC)]
and c.989C>T [A=0.00000
(1/244488, GnomAD)] (P.A330F)
and c.1169G>A
[T=0.00009 (22/244260, GnomAD)] (P.R390H) (Figure 1). These variants were not
detected in any of the 50 healthy controls. In comparison to the previous
studies, R390H mutation was common in Chinese PCG patients[13],
but A330F mutation was novel
in Chinese Han population. It is also important to highlight that it is for the
first time that these two mutations were identified in single PCG patient
worldwide. Furthermore, in addition to these two missense mutations, four other
single-nucleotide polymorphisms (SNPs) were also identified in other analyzed
PCG patients or healthy controls, including c.142C>G (P.R48W), c.355G>T (P.A119S), c.1347T>C
(P.D449D), and c.1294C>G
(P.L432V).
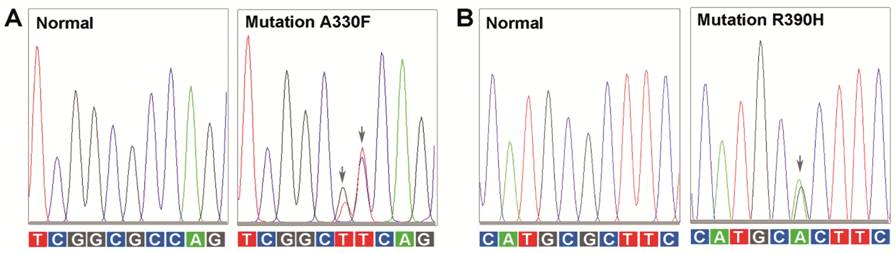
Figure 1
Electropherograms of two mutations in the CYP1B1 gene The normal and the mutated nucleic acids
from region 983 to 992 (A); and region 1164 to 1173 (B) of CYP1B1 gene
have been listed. The mutated nucleic acids are labeled with arrows.
Next, to
predict the functional correlation of these mutations in the CYP1B1
gene, protein structure was analyzed using PyMOL software. As shown
in Figure 2, A330F
and R390H mutations disrupted the orientation of I-helix and K-helix,
respectively. More specifically, the link between the substrate binding region
and the CCS, the torsion of I-helix switched lactamine into phenylalanine in
the side chain of the protein. The benzene ring in the phenylalanine can form the connections with the α-naphthoflavone
(ANF) ligand, and thus
can create stearic hindrance between ANF ligand and heme iron. In addition, another
mutation R390H changed K-helix in CCS, and resulted in the formation of
unstable heme-protein complex. Together, these findings indicated that both A330F and R390H were important alleles to
maintain the fundamental properties of CYP1B1 gene, and might contribute
to severe clinical phenotype.
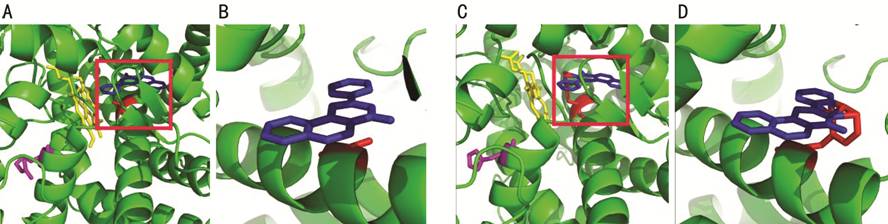
Figure 2 The structural analysis of
wild-type and mutated CYP1B1 protein A: The normal CYP1B1 protein structure
including amino acid, alanine (labeled as red) at position 330 and arginine
(labeled as purple) at position 390. The ANF ligand and HEM (Protoporphyrin Ix
Containing Fe) ligand have been labeled as blue and yellow, respectively. B:
The interaction between alanine and ANF ligand under high magnification. C, D:
The influence of the mutations on the interactions between amino acid and
ligands.
Interestingly, our clinical findings confirmed our
assumption that PCG patient with A330F
and R390H mutations exhibited severe glaucoma phenotype. This patient
specifically had PCG symptoms at birth with photophobia, tearing, and corneal whitening. However,
no family history of PCG has been identified in this patient. As shown in
Figure 3, the IOPs in the right and left eye of this patient were 42 and 43 mm Hg, respectively, at 6-month of age,
and both horizontal corneal diameters were 12 mm.
Goniotomy was performed on both eyes of this
patients at Linyi People’s Hospital, and after the surgery, the reduced IOPs were maintained
for 6mo, but then gradually increased. At the age of 3 years, the IOP of both
eyes reached up to 50-60 mm
Hg. Subsequently, after trabeculotomy, the postoperative IOPs were again
reduced, and maintained for only another 6mo. After the patient turned 11-years-old, the corrected
visions were only 0.07 and 0.05 and the IOPs of his right and left eye reached
to 39 and 36 mm Hg, respectively. Goniotomy along with trabeculotomy
was again performed to reduce his IOPs. Surprisingly, the elevated IOPs could
not be reduced effectively this time. Later glaucoma valve was implanted to his
left eye in the affiliated hospital of Qingdao University, which then led to
reduced IOPs of 16 mm
Hg. Next, with the help of betamethanil eye drops, the IOPs were stabilized at
18-23 mm Hg for the right
eye, and 14-16 mm Hg for the left
eye. More recently, the corrected vision of the right eye was 0.05, and for the
left eye, it was finger counting (10 cm),
with corneal diameter of 13 mm,
for both eyes. The structures behind the iris could not be observed by slit
lamp, due to the adherence of iris in the pupil area to mildly opaque lens.
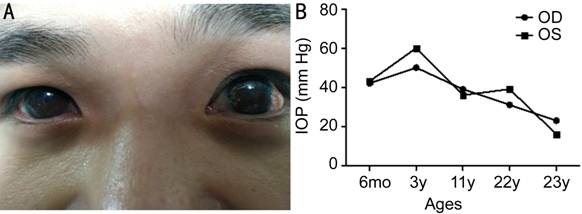
Figure 3 The
clinical characteristics of PCG patient with A330F and R390F
mutations A: The appearance of the eyes at
23 years of age; B: Information about the changes in IOPs after different
treatments and ages.
DISCUSSION
The
congenital glaucoma has mainly been characterized as genetically inherited
disease, and specifically CYP1B1 gene mutations show association with
its pathogenesis[14-17].
Importantly, more than 150 mutations in the CYP1B1 gene have been
reported to be associated with PCG pathophysiology, but recently specific CYP1B1
mutations have been predominantly linked with severe glaucomatous phenotypes[18]. Our study detected common variant, R390H in the CYP1B1
gene among Chinese PCG patient. This mutation has been reported in different
ethnic groups worldwide[19-20],
and has been shown to cause severe angle abnormalities. In addition, our study
also demonstrated the presence of another novel variant, A330F among the same Chinese PCG patient. However, this
additional variant has previously been only reported in Japanese[21] and South Korean[22].
According to the SNP database in NCBI
(https://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=1545), the mutant allele
frequency of A330F
is much lower than the frequency in R390H. The presence of A330F in limited number of PCG patients in
Asia suggest that it is a different allele, capable of inducing severe PCG
phenotypes. Up until this report, no evidence existed about the co-occurrence
of both these two missense mutations, R390H and A330F, in same congenital glaucoma patient. The Chinese
PCG patient in our study was indeed the first one to show this trend in the
world. Further clinical observation confirmed the correlation between the
presence of these mutations and patient displaying severe glaucomatous
phenotypes. Multiple surgeries and combined medicinal treatment was required to
reduce the elevated IOP in this patient.
As the
functional importance of CYP1B1 can be predicted through its protein
structure analysis[23], it can act as an
excellent strategy to understand the correlation between specific mutations and
severe angle abnormalities. The carboxyl terminus of CYP1B1 protein includes
a substrate binding region and CCS, while the N-terminal of CYP1B1
protein includes a membrane-spanning domain and a hinge region. Generally, the
critical mutations in CYP1B1 gene has been located in the hinge region
and CCS[2]. The critical
mutation, R390H
observed in our study usually disrupt the orientation of K-helix in
CCS, which then affects the formation of a stable haemoglobin complex and molecular folding[9,24]. This
mutation clearly changes the fundamental features of CYP1B1, and can
lead to severe developmental abnormalities as reported previously[22]. Another
mutation detected in the CYP1B1 gene in PCG patient in our study was A330F, which is located in the I-helix and
provide link between substrate binding region and CCS. Historically, the
mutation in this region is not as common as R390H, and thus has not been
previously considered very crucial in PCG patients. However, recently more and
more numbers of PCG patients have confirmed the close correlations between
severe phenotypes and this mutation. In addition, mutations like E229K[25] and S239R[26], have also been
shown to involved in the disruption of the three-dimensional structures of
I-helix, and subsequently lead to severe glaucomatous phenotypes. Moreover,
consistent with earlier analysis, our structural analysis also suggested that
the mutation in the I-helix can block binding of substrates with heme iron.
Altogether, these cumulative evidences indicate that the link region is also an
equally important domain for maintaining the function of CYP1B1 gene,
besides the hinge region and CCS.
The CYP1B1
enzyme has been shown to play an important and crucial catalytic role in the
cholesterol, steroids, and other lipids synthesis[27-29]. All these metabolic reactions and products seem to
be important in the differentiation and growth of multiple tissues. In the
context of PCG, CYP1B1 mutations cause significant change in retinol,
which is the key metabolite for the development of trabecular meshwork (TM)[30-31]. Multiple studies have
indicated about the important roles of retinol in the TM development; like
mutations in the retinol receptor gene and the retinoic acid receptor β cause
severe developmental defects[32]. The retinoic
acid receptors and retinoid X
receptors expressed in the TM can transform retinol into its active derivatives
which then regulate the transcription of myocilin gene[29,33]. In addition, the function of the CYP1B1 gene
in regulating aqueous humor outflow is controversial, but glaucoma pathology is
highly related with the mutation and expression of this gene. In our study, we
speculate that heme instability caused by R390H and A330F mutations can lead to the abnormalities in
retinol metabolism, and is supported by the functional studies published in
recent years[30]. Collectively, the evidence
indicates that the retinol metabolic abnormalities due to R390H and A330F mutation might contribute to severe
glaucomatous phenotypes in PCG patients. Alternatively, another major cause of
TM pathogenesis can be abnormal oxidation status due to abnormal CYP1B1
gene activity. Presence of oxidative stress during early development can cause TM hypoplasia,
which then can lead to PCG development[34]. It has been
specifically reported that antioxidant enzymes are deficient in the TM of PCG
patient, and the presence of high H2O2 and superoxide
anions in the aqueous humour can easily facilitate their infiltration into the
TM, and can subsequently
exert oxidative stress, eventually leading to TM pathogenesis[35].
Currently,
goniotomy is the major approach to treat congenital glaucoma. Although the
single surgery can benefit certain PCG patients, but in some patients with
specific mutations in CYP1B1 genes, repetitive surgeries combined with
other medicine are required for treatment. But the combined treatments are also
not effective to reduce IOP in these patients, and therefore, it is necessary
to explore novel approaches for treatment of PCG patients, especially for the
patients like in our study with multiple CYP1B1 mutations. In this
direction, a recent study reported that abnormal cellular function of TM cells
from CYP1B1-/- mouse can be restored by the treatment with
the free radical scavenger, N-acetylcysteine[34].
Similarly, supplementing antioxidant enzymes or/and the retinol to the TM, can
also probably benefit PCG patients with A330F
and R390H mutations. In addition, the CRISPR-Cas9 based gene therapy and stem
cells-based therapy have also been reported as promising approaches to rescue
the glaucomatous phenotypes for open-angle glaucoma through rebuilding damaged
TM[36-37]. Thus, it would be
interesting to test these approaches in PCG patients.
Overall, our
study demonstrated the presence of novel mutations in the Chinese PCG patients.
This is the first report about co-existence of A330F and R390H mutations in the same PCG patient.
Furthermore, the structural changes caused by these two mutations renewed our
current understanding about the role of key domains of CYP1B1 gene in
influencing congenital glaucoma phenotype. It is evident that besides CCS
domain, additional regions can also affect the structure of CCS, and are
crucial in regulating the function of CYP1B1 in terms of causing TM abnormalities. However, future
studies are required to determine the major influence of these mutations on
metabolism and selection of key metabolites, which can probably lead to the
development of potential strategies for PCG therapy.
ACKNOWLEDGEMENTS
We thank Dr. Ke-Wei Wang of Qingdao University and Dr. Li-Ning Lu of
Tsinghua University to support the protein structural analysis study.
Foundation: Supported
by “Clinical medical+X” Project from Department of Medicine of Qingdao
University.
Conflicts of
Interest: Song N,
None; Leng L, None; Yang XJ, None; Zhang YQ, None; Tang
C, None; Chen WS, None; Zhu W, None; Yang X, None.
REFERENCES
|
1 deLuise VP, Anderson DR. Primary
infantile glaucoma (congenital glaucoma). Surv Ophthalmol 1983;28(1):1-19.
https://doi.org/10.1016/0039-6257(83)90174-1
|
|
|
|
2 Zhao Y, Sorenson CM, Sheibani N.
Cytochrome P450 1B1 and primary congenital glaucoma. J Ophthalmic Vis Res
2015;10(1):60-67.
https://doi.org/10.4103/2008-322X.156116
|
|
|
|
|
3 Bouyacoub Y, Ben Yahia S, Abroug N,
Kahloun R, Kefi R, Khairallah M, Abdelhak S. CYP1B1 gene mutations causing
primary congenital glaucoma in Tunisia. Ann Hum Genet 2014;78(4):255-263.
https://doi.org/10.1111/ahg.12069
|
|
|
|
|
4 Faiq M, Sharma R, Dada R, Mohanty K,
Saluja D, Dada T. Genetic, biochemical and clinical insights into primary
congenital glaucoma. J Curr Glaucoma Pract 2013;7(2):66-84.
https://doi.org/10.5005/jp-journals-10008-1140
|
|
|
|
|
5 Sarfarazi M, Akarsu AN, Hossain A,
Turacli ME, Aktan SG, Barsoum-Homsy M, Chevrette L, Sayli BS. Assignment of a
locus (GLC3A) for primary congenital glaucoma (Buphthalmos) to 2p21 and
evidence for genetic heterogeneity. Genomics 1995;30(2):171-177.
https://doi.org/10.1006/geno.1995.9888
|
|
|
|
|
6 Akarsu AN, Turacli ME, Aktan SG,
Barsoum-Homsy M, Chevrette L, Sayli BS, Sarfarazi M. A second locus (GLC3B)
for primary congenital glaucoma (Buphthalmos) maps to the 1p36 region. Hum
Mol Genet 1996;5(8):1199-1203.
https://doi.org/10.1093/hmg/5.8.1199
|
|
|
|
|
7 Narooie-Nejad M, Paylakhi SH, Shojaee S,
Fazlali Z, Rezaei Kanavi M, Nilforushan N, Yazdani S, Babrzadeh F, Suri F,
Ronaghi M, Elahi E, Paisán-Ruiz C. Loss of function mutations in the gene
encoding latent transforming growth factor beta binding protein 2, LTBP2,
cause primary congenital glaucoma. Hum Mol Genet 2009;18(20):3969-3977.
https://doi.org/10.1093/hmg/ddp338
|
|
|
|
|
8 Wang A, Savas U, Stout CD, Johnson EF.
Structural characterization of the complex between α-naphthoflavone and human
cytochrome P450 1B1. J Biol Chem 2011;286(7):5736-5743.
https://doi.org/10.1074/jbc.M110.204420
|
|
|
|
|
9 Stoilov I, Akarsu AN, Alozie I, Child A,
Barsoum-Homsy M, Turacli ME, Or M, Lewis RA, Ozdemir N, Brice G, Aktan SG,
Chevrette L, Coca-Prados M, Sarfarazi M. Sequence analysis and homology
modeling suggest that primary congenital glaucoma on 2p21 results from
mutations disrupting either the hinge region or the conserved core structures
of cytochrome P4501B1. Am J Hum Genet 1998;62(3):573-584.
https://doi.org/10.1086/301764
|
|
|
|
|
10 Hollander DA, Sarfarazi M,
Stoilov I, Wood IS, Fredrick DR, Alvarado JA. Genotype and phenotype
correlations in congenital glaucoma. Trans Am Ophthalmol Soc
2006;104:183-195.
|
|
|
|
|
11 Li N, Zhou Y, Du L, Wei M, Chen
X. Overview of cytochrome P450 1B1 gene mutations in patients with primary
congenital glaucoma. Exp Eye Res 2011;93(5):572-579.
https://doi.org/10.1016/j.exer.2011.07.009
|
|
|
|
|
12 Yuan R, Zhao J, Zhao Y, Ma
X, Dai Q, Wang B. Systematic review of CYP1B1 gene mutations in patients with
primary congenital glaucoma in China. J Evid Based Med 2015;1:42-47.
|
|
|
|
|
13 Yang M, Guo X, Liu X, Shen H,
Jia X, Xiao X, Li S, Fang S, Zhang Q. Investigation of CYP1B1 mutations in
Chinese patients with primary congenital glaucoma. Mol Vis 2009;15:432-437.
|
|
|
|
|
14 Faiq M, Mohanty K, Dada R,
Dada T. Molecular diagnostics and genetic counseling in primary congenital
glaucoma. J Curr Glaucoma Pract 2013;7(1):25-35.
https://doi.org/10.5005/jp-journals-10008-1140
|
|
|
|
|
15 Della Paolera M, de Vasconcellos
JP, Umbelino CC, Kasahara N, Rocha MN, Richeti F, Costa VP, Tavares A, de
Melo MB. CYP1B1 gene analysis in primary congenital glaucoma Brazilian
patients: novel mutations and association with poor prognosis. J Glaucoma
2010;19(3):176-182.
https://doi.org/10.1097/IJG.0b013e3181a98bae
|
|
|
|
|
16 Chouiter L, Nadifi S.
Analysis of CYP1B1 gene mutations in patients with primary congenital
glaucoma. J Pediatr Genet 2017;6(4):205-214.
https://doi.org/10.1055/s-0037-1602695
|
|
|
|
|
17 Sitorus R, Ardjo SM, Lorenz
B, Preising M. CYP1B1 gene analysis in primary congenital glaucoma in Indonesian
and European patients. J Med Genet 2003;40(1):e9.
https://doi.org/10.1136/jmg.40.1.e9
|
|
|
|
|
18 Chen Y, Jiang D, Yu L, Katz
B, Zhang K, Wan B, Sun X. CYP1B1 and MYOC mutations in 116 Chinese patients
with primary congenital glaucoma. Arch Ophthalmol 2008;126(10):1443-1447.
https://doi.org/10.1001/archopht.126.10.1443
|
|
|
|
|
19 Tanwar M, Dada T, Sihota R,
Das TK, Yadav U, Dada R. Mutation spectrum of CYP1B1 in North Indian
congenital glaucoma patients. Mol Vis 2009;15:1200-1209.
|
|
|
|
|
20 Rauf B, Irum B, Kabir F,
Firasat S, Naeem MA, Khan SN, Husnain T, Riazuddin S, Akram J, Riazuddin SA.
A spectrum of CYP1B1 mutations associated with primary congenital glaucoma in
families of Pakistani descent. Hum Genome Var 2016;3:16021.
https://doi.org/10.1038/hgv.2016.21
|
|
|
|
|
21 Ohtake Y, Tanino T, Suzuki
Y, Miyata H, Taomoto M, Azuma N, Tanihara H, Araie M, Mashima Y. Phenotype of
cytochrome P4501B1 gene (CYP1B1) mutations in Japanese patients with primary
congenital glaucoma. Br J Ophthalmol 2003;87(3):302-304.
https://doi.org/10.1136/bjo.87.3.302
|
|
|
|
|
22 Kim HJ, Suh W, Park SC, Kim
CY, Park KH, Kook MS, Kim YY, Kim CS, Park CK, Ki CS, Kee C. Mutation
spectrum of CYP1B1 and MYOC genes in Korean patients with primary congenital
glaucoma. Mol Vis 2011;17:2093-2101.
|
|
|
|
|
23 Chavarria-Soley G, Sticht H,
Aklillu E, Ingelman-Sundberg M, Pasutto F, Reis A, Rautenstrauss B. Mutations
in CYP1B1 cause primary congenital glaucoma by reduction of either activity
or abundance of the enzyme. Hum Mutat 2008;29(9):1147-1153.
https://doi.org/10.1002/humu.20786
|
|
|
|
|
24 Su CC, Liu YF, Li SY, Yang
JJ, Yen YC. Mutations in the CYP1B1 gene may contribute to juvenile-onset
open-angle glaucoma. Eye (Lond) 2012;26(10):1369-1377.
https://doi.org/10.1038/eye.2012.159
|
|
|
|
|
25 Hollander DA, Sarfarazi M,
Stoilov I, Wood IS, Fredrick DR, Alvarado JA. Genotype and phenotype
correlations in congenital glaucoma: CYP1B1 mutations, goniodysgenesis, and
clinical characteristics. Am J Ophthalmol 2006;142(6):993-1004.
https://doi.org/10.1016/j.ajo.2006.07.054
|
|
|
|
|
26 Achary MS, Reddy AB,
Chakrabarti S, Panicker SG, Mandal AK, Ahmed N, Balasubramanian D, Hasnain
SE, Nagarajaram HA. Disease-causing mutations in proteins: structural
analysis of the CYP1B1 mutations causing primary congenital glaucoma in
humans. Biophys J 2006;91(12):4329-4339.
https://doi.org/10.1529/biophysj.106.085498
|
|
|
|
|
27 Choudhary D, Jansson I,
Sarfarazi M, Schenkman JB. Characterization of the biochemical and structural
phenotypes of four CYP1B1 mutations observed in individuals with primary congenital
glaucoma. Pharmacogenet Genomics 2008;18(8):665-676.
https://doi.org/10.1097/FPC.0b013e3282ff5a36
|
|
|
|
|
28 Kaur K, Mandal AK, Chakrabarti
S. Primary congenital glaucoma and the involvement of CYP1B1. Middle East Afr
J Ophthalmol 2011;18(1):7-16.
https://doi.org/10.4103/0974-9233.75878
|
|
|
|
|
29 Vasiliou V, Gonzalez FJ.
Role of CYP1B1 in glaucoma. Annu Rev Pharmacol Toxicol 2008;48(1):333-358.
https://doi.org/10.1146/annurev.pharmtox.48.061807.154729
|
|
|
|
|
30 Prat C, Belville C, Comptour
A, Marceau G, Clairefond G, Chiambaretta F, Sapin V, Blanchon L. Myocilin
expression is regulated by retinoic acid in the trabecular meshwork-derived
cellular environment. Exp Eye Res 2017;155:91-98.
https://doi.org/10.1016/j.exer.2017.01.006
|
|
|
|
|
31 Teixeira LB, Zhao Y,
Dubielzig RR, Sorenson CM, Sheibani N. Ultrastructural abnormalities of the
trabecular meshwork extracellular matrix in Cyp1b1-deficient mice. Vet Pathol
2015;52(2):397-403.
https://doi.org/10.1177/0300985814535613
|
|
|
|
|
32 Williamson KA, FitzPatrick DR.
The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur J
Med Genet 2014;57(8):369-380.
https://doi.org/10.1016/j.ejmg.2014.05.002
|
|
|
|
|
33 Banerjee A, Chakraborty S,
Chakraborty A, Chakrabarti S, Ray K. Functional and structural analyses of
CYP1B1 variants linked to congenital and adult-onset glaucoma to investigate
the molecular basis of these diseases. PLoS One 2016;11(5):e0156252.
https://doi.org/10.1371/journal.pone.0156252
|
|
|
|
|
34 Zhao Y, Wang SJ, Sorenson
CM, Teixeira L, Dubielzig RR, Peters DM, Conway SJ, Jefcoate CR, Sheibani N. Cyp1b1
mediates periostin regulation of trabecular meshwork development by
suppression of oxidative stress. Mol Cell Biol 2013;33(21):4225-4240.
https://doi.org/10.1128/MCB.00856-13
|
|
|
|
|
35 Tanwar M, Dada T, Sihota R,
Dada R. Mitochondrial DNA analysis in primary congenital glaucoma. Mol Vis
2010;16:518-533.
|
|
|
|
|
36 Choudhary D, Jansson I,
Schenkman JB. CYP1B1, a developmental gene with a potential role in glaucoma therapy.
Xenobiotica 2009;39(8): 606-615.
https://doi.org/10.1080/00498250903000198
|
|
|
|
|
37 Bolinches-Amorós A, Lukovic D,
Castro AA, León M, Kamenarova K, Kaneva R, Jendelova P, Blanco-Kelly F, Ayuso
C, Cortón M, Erceg S. Generation of a human iPSC line from a patient with
congenital glaucoma caused by mutation in CYP1B1 gene. Stem Cell Res
2018;28:96-99.
https://doi.org/10.1016/j.scr.2018.01.004
|
|