
Citation:
Hu YS, Song H, Li Y, Xiao ZY, Li T. Whole-exome sequencing identifies novel
mutations in genes responsible for retinitis pigmentosa in 2 nonconsanguineous
Chinese families. Int J Ophthalmol
2019;12(6):915-923
DOI:10.18240/ijo.2019.06.06
・Basic
Research・
Whole-exome
sequencing identifies novel mutations in genes responsible for retinitis
pigmentosa in 2 nonconsanguineous Chinese families
Yan-Shan Hu, Hui Song, Yin Li, Zi-Yun Xiao, Tuo Li
Department
of Ophthalmology, the Central Hospital of Enshi Autonomous Prefecture, Enshi
Clinical College of Wuhan University, Enshi 445000, Hubei Province, China
Correspondence
to: Tuo Li.
Department of Ophthalmology, the Central Hospital of Enshi Autonomous
Prefecture, Enshi Clinical College of Wuhan University, Enshi 445000, Hubei
Province, China. 13986840088@139.com
Received: 2018-08-18
Accepted: 2018-09-25
Abstract
AIM: To
detect the pathogenetic mutations responsible for nonsyndromic autosomal
recessive retinitis pigmentosa (RP) in 2 nonconsanguineous Chinese families.
METHODS: The clinical data, including detailed medical history, best corrected
visual acuity (BCVA), slit-lamp biomicroscope examination, fundus photography, optical
coherence tomography, static perimetry, and full field electroretinogram, were
collected from the members of 2 nonconsanguineous Chinese families
preliminarily diagnosed with RP. Genomic DNA was extracted from the probands
and other available family members; whole-exome sequencing was conducted with
the DNA samples provided by the probands, and all mutations detected by
whole-exome sequencing were verified using Sanger sequencing in the probands
and the other available family members. The verified novel mutations were
further sequenced in 192 ethnicity matched healthy controls.
RESULTS: The patients from the 2 families exhibited the
typical symptoms of RP, including night blindness and progressive constriction
of the visual field, and the fundus examinations showed attenuated retinal
arterioles, peripheral bone spicule pigment deposits, and waxy optic discs.
Whole-exome sequencing revealed a novel nonsense mutation in FAM161A (c.943A>T, p.Lys315*) and compound heterozygous
mutations in RP1L1 (c.56C>A, p.Pro19His; c.5470C>T, p.Gln1824*). The
nonsense c.5470C>T,
p.Gln1824* mutation was novel. All mutations were verified by Sanger
sequencing. The mutation p.Lys315* in FAM161A co-segregated with the phenotype, and all the
nonsense mutations were absent from the ethnicity matched healthy controls and
all available databases.
CONCLUSION: We identify 2 novel mutations in genes responsible
for autosomal recessive RP, and the mutation in FAM161A is reported for the first time in a Chinese
population. Our result not only enriches the knowledge of the mutation
frequency and spectrum in the genes responsible for nonsyndromic RP but also
provides a new target for future gene therapy.
KEYWORDS: retinitis pigmentosa; nonsyndromic;
whole-exome sequencing; mutation; novel
DOI:10.18240/ijo.2019.06.06
Citation:
Hu YS, Song H, Li Y, Xiao ZY, Li T. Whole-exome sequencing identifies novel
mutations in genes responsible for retinitis pigmentosa in 2 nonconsanguineous
Chinese families. Int J Ophthalmol
2019;12(6):915-923
Outline
INTRODUCTION
SUBJECTS
AND METHODS
RESULTS
DISCUSSION
ACKNOWLEDGEMENTS
REFERENCES
INTRODUCTION
Retinitis pigmentosa (RP) is a group of heterogenous
hereditary retinal degeneration diseases, which are characterized by the
progressive loss of function in photosensory cells and pigment epithelium[1-2]. RP can
manifest in a syndromic or a nonsyndromic form and is typically characterized
by night blindness, progressive constriction of the visual field, changes in the fundus and
abnormal electroretinogram results. The incidence of nonsyndromic RP worldwide
is approximately 1/5000-1/3000. At present, it is estimated that there were
approximately 2.5 million RP patients in the world, and RP is an important
cause of irreversible blindness due to fundus disease[3-4]. In China,
the incidence is approximately 1/3800[5], which
means that there are approximately 370 000 patients in China suffering from
nonsyndromic RP.
Genetic predisposition plays an important role in the
pathogenesis of RP, and RP can be inherited as an autosomal dominant (AD),
autosomal recessive (AR) or X-linked trait. Currently, there are 92 genes listed
in RetNet (available in the public domain at https://sph.uth.edu/retnet/)
that have been identified as being involved in the development of RP. These
genes are involved in the transduction cascade of the optical signal and the
regulation of the transcription and translation of other retinal genes. It has
been reported that these genes account for only 60% of the nonsyndromic RP
patients[6-8]; therefore,
more efforts should be made to uncover the genetic basis of RP.
Whole-exome sequencing (WES) has been proven to be an
efficient method to detect the genetic defects underlying hereditary diseases,
and it primarily captures information about the coding regions of genes,
providing a faster and more efficient way to explore the genetic causes of
hereditary diseases[9-10]. In this
study, WES was adopted to uncover the disease-causing mutations in 2
nonconsanguineous Chinese families with nonsyndromic RP.
SUBJECTS AND METHODS
Ethical Approval
The current study was approved by the Ethics Committee of
The Central Hospital of Enshi Autonomous Prefecture and adhered to the tenets
of the Declaration of Helsinki. Written informed consent was obtained from all
the probands and their family members (or their guardians if they were minors).
Subjects and DNA Specimens Two
nonconsanguineous families with RP and 192 ethnicity matched healthy controls
were recruited in the Ophthalmologic Centre of the Central Hospital of Enshi
Autonomous Prefecture in Southwest China. The diagnosis of nonsyndromic RP was
made based on the patients’ medical histories, symptoms and physical
examinations. All the patients underwent autorefraction (Topcon KR-8000,
Paramus, NJ, USA), subjective optometry, slit-lamp biomicroscopic examination,
IOL master (Carl Zeiss Meditec AG, Jena, Germany), fundus photography (Canon
CF-60UD, Tokyo, Japan), optical coherence tomography (Heidelberg Engineering
HRA+OCT, Heidelberg, Germany), fundus autofluorescence (Cirrus HD-OCT 4000,
Carl Zeiss Meditec Inc., Jena, Germany), static perimetry (Humphrey Field
Analyser, Carl Zeiss Meditec Inc., Dublin, CA, USA), and full field
electroretinography (Roland Electrophysiological Test Unit RETI-Scan 21, Roland
Consult, Berlin, Germany). The electroretinogram was conducted in accordance
with the standards of the International Society for Clinical Electrophysiology
of Vision[11]. DNA specimens were collected from peripheral
venous blood using a blood DNA extraction kit (Promega, Madison, Wisconsin,
USA) according to the manufacturer’s instructions and stored in TE buffer.
Whole-exome Sequencing WES was
conducted with the DNA samples from the probands by a commercial service
(Macrogen Inc., Seoul, Korea). The genomic DNA of each proband was enriched
with an Agilent SureSelect Human All Exon Enrichment Kit V5 array (Agilent
Technologies, Santa Clara, CA, USA), and the enriched DNA fragments were
sequenced with an Illumina HiSeq4000 system (Illumina, Santiago, CA, USA). The
sequencing depth of every sample was greater than 125-fold. Burrows Wheeler
Aligner software was used to map short reads to the hg19 reference genome
(available in the public domain at http://bio-bwa.sourceforge.net/bwa.shtml). Variant
calling and filtering was conducted with GATK (Genome Analysis Toolkit)
software (available in the public domain at
https://www.broadinstitute.org/gatk/). Variant annotation was performed with
SnpEff (available in the public domain at http://snpeff.sourceforge.net/SnpEff.html).
Data Analysis WES data were selected for the known
causative genes of nonsyndromic RP. Variants in these genes were filtered by
the following criteria: 1) those with a minor allele frequency (MAF) greater
than 0.005 in the
1000 Genomes database (available in the public domain at
http://www.internationalgenome.org/), the Exome Aggregation Consortium database
(ExAC, available in the public domain at http://exac.broadinstitute.org/), the
Genome Aggregation database (gnomAD, available in the public domain at
http://gnomad.broadinstitute.org/), the NHLBI GO Exome Sequencing Project
database (ESP, available in the public domain at http://evs.gs.washington.edu/)
and the database of single nucleotide polymorphisms (dbSNP, available in the
public domain at https://www.ncbi.nlm.nih.gov/snp) were filtered out; 2)
variants located in the intron region that did not affect the splicing site
were filtered out; 3) synonymous variants that did not affect the splicing site
were filtered out; and 4) variants predicted to be benign or tolerated by
Polymorphism Phenotyping v2 (PolyPhen2, available in the public domain at
http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant From Tolerant (SIFT,
available in the public domain at http://sift.jcvi.org/) or
PROVEN (available in the public domain at https://provean.jcvi.org/index.php)
were filtered out. After the data were filtered, only nonsynonymous variants
remained for further verification.
Sanger Sequencing for Variant Verification and Segregation
Analysis The
following verification and segregation analyses were conducted on the mutations
that remained after the data were filtered. Polymerase chain reaction (PCR) was
carried out to amplify the fragments containing the variants. Primers were
designed with the Primer3 online website (available in the public domain at
http://primer3.ut.ee/), and all the primers are listed in Table 1. The
amplicons were sequenced with an ABI BigDye Terminator v3.1 Cycle Sequencing
kit using an ABI 3100 and a 3500xL Dx Genetic Analyser (Applied Biosystems,
Foster City, CA, USA). The genomic DNA reference sequences were downloaded from
the NCBI GenBank (available in the public domain at
https://www.ncbi.nlm.nih.gov/), and the sequencing data were analysed using the
SeqMan II programme of the Lasergene software package (DNAStar Inc., Madison,
WI, USA). The DNA samples of all the probands and their available family
members were Sanger sequenced, and the segregation analysis was conducted in
accordance with the respective inheritance mode. Further verification was
carried out in 192 ethnicity matched healthy controls. The amino acid sequences
of different species were acquired from the NCBI website (available in the
public domain at https://www.ncbi.nlm.nih.gov/), and the conservation analysis
was conducted using the MegAlign programme of the Lasergene software package
(DNAStar Inc., Madison, WI, USA).
Table 1 PCR primers used for FAM161A and RP1L1
|
Gene
|
Mutation
|
Sequence
|
Primer length (bp)
|
Amplicon size (bp)
|
|
FAM161A
|
c.943A>T
|
F: 5’-TGGACAGACTTTTGTGTTGAGG-3’
|
22
|
689
|
|
|
|
R: 5’-TCAAAATCAGGAGTTGGGCAC-3’
|
21
|
|
|
RP1L1
|
c.56C>A
|
F: 5’-GCACCTCTAGAAAGACGGGA-3’
|
20
|
291
|
|
|
|
R: 5’-GGCGCTGAAGGTCTTAAAGG-3’
|
20
|
|
|
|
c.5470C>T
|
F: 5’-GAGACAAAGATCCCAAACTCGG-3’
|
22
|
698
|
|
|
|
R: 5’-GGTCTCCACTTCAACCTCCA-3’
|
20
|
|
RESULTS
Clinical Manifestations and Pedigree Information In the
current study, we investigated 2 nonsyndromic RP families, EQT33 and EQT38. The
proband of EQT33 was a 19-year-old male whose chief complaint was night
blindness with decreased vision. The main complaint was decreased dark
adaptation 13 years ago, and over the past 13y, his visual acuity slowly
decreased. When he first visited our ophthalmologic centre, his anterior ocular
segment was normal and BCVA revealed that he could perceive hand movements with
both eyes. The mydriasis fundus examination demonstrated peripheral pigment
bone spicule deposits, slightly waxy optic discs, attenuated retinal arterioles
and macular degeneration in both eyes. The OCT showed blurring of inner and
outer segment layers (IS/OS). The electroretinogram (ERG) was severely reduced
in all six tests (Figure 1). His visual field was not measured due to his poor
visual acuity. The proband’s younger sister (EQT33 II-2) also suffered from RP,
but with milder symptoms. She had sensed a decrease in dark adaptation at the
age of 10, and her BCVA was 0.15 for the right eye and 0.12 for the left at her
first visit. The mydriasis fundus examination showed slightly attenuated
retinal arterioles, a small number of retinal pigment deposits in the
peripheral retina and tigroid fundus. Other family members did not exhibit any
symptoms of RP, and their physical examinations revealed normal results for
both eyes; therefore, we concluded that the most likely mode of inheritance in
EQT33 was AR. The clinical information for EQT33 is summarized in Table 2.
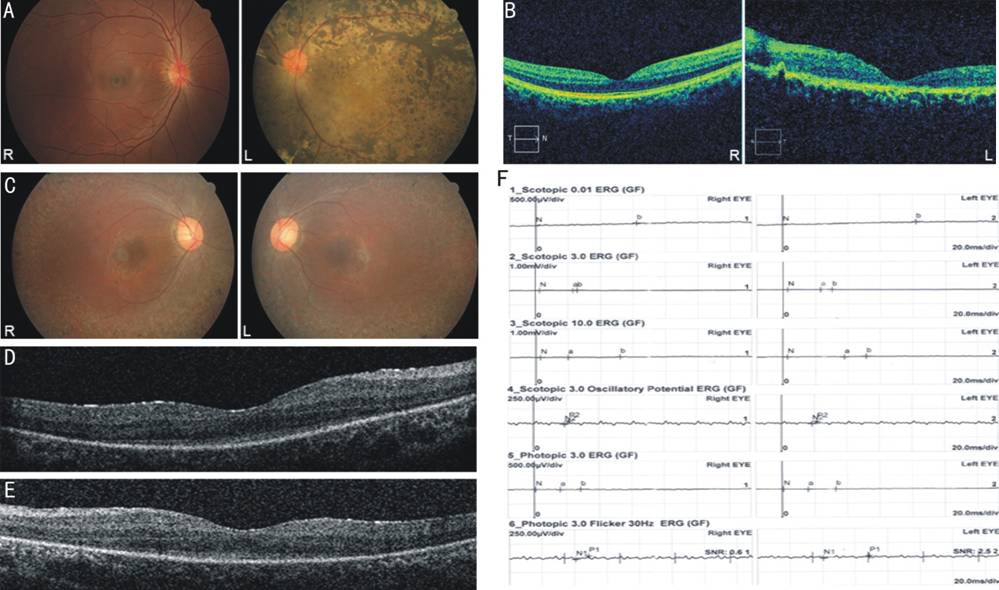
Figure 1 Ophthalmic examinations of
the probands from two RP families A: Fundus photograph of the proband
in EQT38. Fundus photograph showed panretinal dense pigment deposits, waxy
optic disc, attenuated retinal arterioles and macular degeneration in the left
eye, while the retina of her right eye was normal. B: OCT image of the
proband in EQT38. OCT of the macula in the left eye exhibited marked thinning and
blurring of all retinal layers, while the OCT in the right eye was normal. C:
Fundus photograph of the proband in EQT33. Fundus photograph showed peripheral
pigment bone spicule deposits, slightly waxy optic discs, attenuated retinal
arterioles and macular degeneration in both eyes. D, E: OCT image of the
proband in EQT33. OCT image of the maculae in both eyes showed blurring of the
inner and outer segment layers (IS/OS). D: Right eye; E: Left eye. F: ERG of
the proband in EQT33. The ERG was severely deduced in all six tests.
Table 2 Clinical information regarding probands in the
EQT33 and EQT38 families
|
Family No.
|
Gender
|
Age at exam (y)
|
Refraction error, OD/OS (D)
|
BCVA, OD/OS
|
Fundus,
OD/OS
|
OCT,
OD/OS
|
ERG,
OD/OS
|
Visual field,
OD/OS
|
|
EQT33
|
F
|
20
|
+0.5/+0.75
|
HM/HM
|
Affected/affected
|
MA/MA
|
RRCS/RRCS
|
NA/NA
|
|
EQT33II-2
|
M
|
19
|
-11.5/-11.0
|
0.15/0.12
|
Affected/affected
|
MA/MA
|
RRCS/RRCS
|
TVF/TVF
|
|
EQT33I-1
|
F
|
50
|
0/0
|
1.0/1.0
|
NA/NA
|
NA/NA
|
NA/NA
|
NA/NA
|
|
EQT33I-2
|
M
|
50
|
-0.5/-0.5
|
0.8/0.8
|
NA/NA
|
Normal/normal
|
NA/NA
|
NA/NA
|
|
EQT38
|
F
|
26
|
-3.75/+0.5
|
0.8/HM
|
Normal/affected
|
Normal/MT
|
NA/NA
|
Normal/NA
|
BCVA: Best corrected visual acuity; D: Diopter; ERG:
Electroretinogram; OD: Right eye; OS: Left eye; HM: Hand movement; MA: Macular
atrophy; MT: Macular thinning; TVF: Tubular visual field; RRCS: Reduced rod and
cone response; NA: Not available.
The proband EQT38 was a 26-year-old female. Her left eye
had suffered from poor visual acuity since birth, and for as long as she could
remember, she had been unable to see anything at night with her left eye. In
the last 26y, her left eye gradually developed exotropia. Unlike her left eye,
her right eye had a normal visual acuity. When she first came to our
ophthalmologic centre, her BCVA was 0.8 for her right eye and hand movements
for her left eye. Slit-lamp microscopic examination revealed normal anterior
segments for both eyes. The fundus examination demonstrated severe signs of RP
in the left eye, including panretinal dense pigment deposits (in some places,
the pigments connected to form flakes), waxy optic discs, attenuated retinal
arterioles and macular degeneration. Interestingly, the retina of her right eye
was normal without any sign of pigment deposits, waxy optic disc or retinal
arterioles. The macular OCT of her left eye exhibited marked thinning and
blurring of all retina layers, while the OCT of her right eye was normal. The
visual field was not measured in her left eye due to the poor BCVA, and the
visual field in her right eye was normal. She stated that her parents did not
suffer from ocular diseases, and her child did not exhibit any signs of night
blindness or poor visual acuity; therefore, we speculated that the inheritance
mode in this pedigree was also AR. Unfortunately, due to economic difficulties,
she did not return to our centre for follow-up, so we could not perform an ERG
to generate more evidence supporting the diagnosis.
Whole-exome Sequencing Results On average,
8.7 billion read bases were obtained from the WES for EQT33 and EQT38, and the
number of total reads was 86.2 million per sample; 97.0% of the read bases
reached Q20, while 92.9% of the read bases reached Q30. The throughput depth of
target regions reached 172.7×. After being mapped to the human reference genome
sequence (hg19), the mean depth of target regions reached 92.6×. Then the base
quality score recalibration, indel realignment, duplicate removal, SNP and
INDEL discovery and genotyping were conducted, and we obtained 82783 variants.
After the filtration described in the Methods section, a total of 478 variants
remained for further analysis. The WES results are summarized in Table 3, and
the histogram of the depth distribution in target regions is shown in Figure 2.
Then, we mapped these 478 variants to the genes responsible for nonsyndromic RP
listed in RetNet. Ultimately, we obtained a homozygous nonsense mutation in FAM161A (c.943A>T, p.Lys315*) in EQT33, and a compound
heterozygous mutation in RP1L1
(c.56C>A,
p.Pro19His; c.5470C>T, p.Gln1824*)
in EQT38. The nonsense mutation p.Lys315* in FAM161A and p.Gln1824* in RP1L1 were not included in the 1000 Genomes database,
ExAC, gnomAD, ESP, dbSNP or Human Gene Mutation Database (HGMD, available in
the public domain at http://www.hgmd.cf.ac.uk/ac/index.php), and no
published papers have reported these 2 mutations. The missense mutation
p.Pro19His in RP1L1 has an
allele frequency of 0.0005243 in
East Asian populations in gnomAD. The effect of the amino acid substitution in
the missense (c.56C>A,
p.Pro19His) mutation in RP1L1
was predicted using SIFT, PolyPhen2 and Proven, and all the tools predicted a
damaging impact of the mutation on the protein function. The detailed
information is summarized in Table 4. Conservation analysis demonstrated that
all 3 mutations are highly conserved. The Conservation analysis result is shown
in Figure 3.
Table 3 Summary of WES results
|
Sample
|
Total read bases (bp)
|
Total reads
|
On-target reads
|
Q20 (%)
|
Q30 (%)
|
Coverage
>30× (%)
|
Mean depth of target
regions (×)
|
Total SNP
|
Synonymous variant
|
|
EQT33
|
8246785744
|
81651344
|
56539425
|
95.9
|
90.5
|
90.3
|
95.6
|
81401
|
11390
|
|
EQT38
|
9163448412
|
90727212
|
54079044
|
98.1
|
95.3
|
90.3
|
89.6
|
84166
|
11481
|
Table 4 Mutations detected in the EQT33 and EQT38
pedigrees
|
Family
|
Gene
|
Position
|
Exon
|
DNA change
|
Protein change
|
Status
|
Mutation type
|
Note
|
Allele frequency in control
Poly-Phen2 SIFT proven
|
|
EQT33
|
FAM161A
|
62067196
|
3
|
c.943A>T
|
p.Lys315*
|
HOM
|
Nonsense
|
Novel
|
0/192, NA, NA, NA
|
|
EQT38
|
RP1L1
|
10480656
|
2
|
c.56C>A
|
p.Pro19His
|
HET
|
Missense
|
Reporteda
|
NA, probably, damaging,
damaging, deleterious
|
|
|
RP1L1
|
10466138
|
4
|
c.5470C>T
|
p.Gln1824*
|
HET
|
Nonsense
|
Novel
|
0/192, NA, NA, NA
|
HOM: Homozygous; HET: Heterozygous; NA: Not available. aThe
missense mutation p.Pro19His in RP1L1
has an allele frequency of 0.0005243 in
East Asian populations in gnomAD.
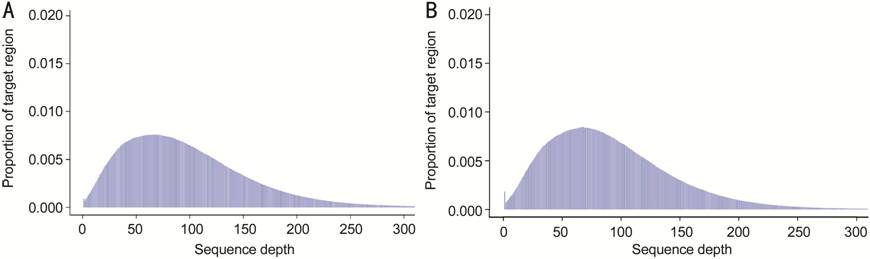
Figure 2 Histogram of the depth distribution in target
regions On average,
8.7 billion read bases were obtained from WES for EQT33 (A) and EQT38 (B), and
the number of total reads was 86.2 million per sample; 97.0% of the read bases
reached Q20, while 92.9% of the read bases reached Q30. The throughput depth of
target regions reached 172.7×.

Figure 3 Amino acid sequences alignment results in
different species A, B:
The p.Lys315* mutation in FAM161A
and p.Pro19His in RP1L1 is highly
conserved in different species; C: The p.Gln1824* mutation in RP1L1 is highly conserved in primates.
Sanger Sequencing for Mutation Verification and
Segregation Analysis To verify
the mutations detected by WES, Sanger sequencing was performed with the samples
from the probands and their available family members. The results demonstrated
the co-segregation of the mutation with the disease phenotype in the EQT33
pedigree. The proband of the EQT38 pedigree did not return for follow-up due to
economic difficulties, and therefore we could not perform the segregation
analysis. The segregation analysis results and the family tree are shown in Figure
4. We next screened 192 ethnicity matched healthy controls for the 2 novel
mutations (c.943A>T,
p.Lys315* and c.5470C>T, p.Gln1824*)
by Sanger sequencing, and neither mutation was detected in the control group.
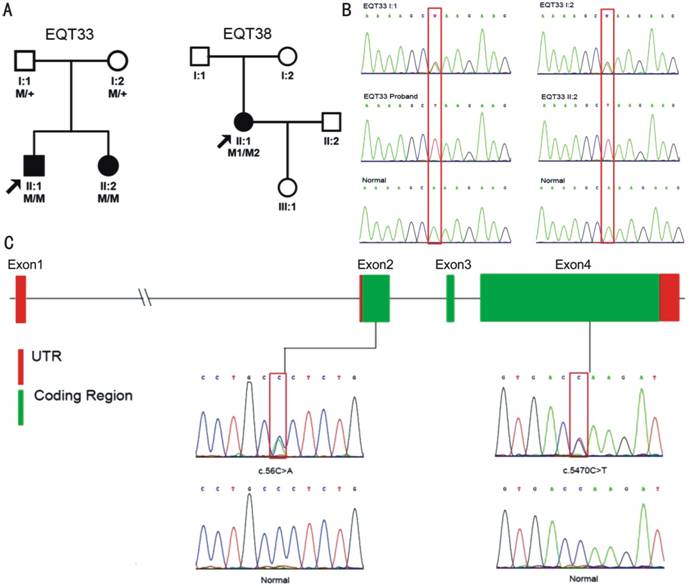
Figure 4 The segregation analysis results
and the family tree A: RP pedigrees presented in this study. The probands
are indicated by the black arrows. +: Wild-type; M: Mutation. B: Mutation
verification of the FAM161A
gene in the EQT33 family. Sanger sequencing demonstrated that the mutation c.943A>T in FAM161A co-segregated with the phenotype. The
proband’s normal parents (EQT33 I:1 and EQT33 I:2) carried heterozygous c.943A>T mutations in FAM161A, while the affected proband and his
affected sister (EQT33 II:2) harbored homozygous c.943A>T mutations in FAM161A. C: Genomic structure of exons encoding the open
reading frame of RP1L1. Three
out of four exons are translated (green), while exon 1, portions of exon 2 and
exon 4 are untranslated (red). The Sanger sequencing results of the compound
heterozygous mutations in RP1L1
(c.56C>A, p.Pro19His; c.5470C>T, p.Gln1824*) are
shown.
DISCUSSION
RP is a group of heterogenous hereditary retinal degeneration
diseases, characterized by the progressive loss of function of the photosensory
cells and pigment epithelium. In the current study, we investigated 2 families
with nonsyndromic AR RP, pedigrees EQT33 and EQT38. All the patients presented
with typical RP symptoms and signs, namely night blindness with decreased
vision, peripheral pigment bone spicule deposits revealed by a mydriasis fundus
examination, waxy optic discs, attenuated retinal arterioles and macular
degeneration. It is worth noting that the proband in EQT38 had suffered from RP
in only the left eye. WES and the subsequent Sanger sequencing identified a
novel nonsense mutation in FAM161A
(c.943A>T,
p.Lys315*) and compound heterogeneous mutations (c.56C>A, p.Pro19His; c.5470C>T, p.Gln1824*), of which c.5470C>T, p.Gln1824* was determined to be
novel.
FAM161A
was first reported to cause nonsyndromic RP by Langmann et al[12] in an
Indian population in 2010; since then, several articles have reported a few more
mutations in different ethnicities[13-15]. The
mutation frequency of FAM161A
in North America is approximately 1%[13]. Van Schil et
al[14] concluded that mutations in FAM161A were responsible for 2% of AR RP cases
in the Dutch and Belgian populations, while Bandah-Rozenfeld et al[15] determined
that mutations in FAM161A
were responsible for approximately 12% of AR RP families in a cohort from
Israel and Palestine. To date, mutations in FAM161A have not been reported to cause RP in the Chinese
population; thus, our study is the first concerning the role of mutations in
FAM161A in the
development of RP in a Chinese population.
Although most of the mutations detected so far have been
nonsense mutations located in exon 3, the largest exon in FAM161A[16-17], the RP phenotypes
differ in distinct populations, even within the same family[18-21]. In North
America, Venturini et al[13] observed
that patients with FAM161A
mutations exhibit early-onset RP with relatively good visual acuity and greatly
reduced cone response on ERG tests, while Bandah-Rozenfeld et al[15] reported
extinguished rod-cone ERG responses in a majority of their patients. Other
researchers found that some RP patients have cataracts or myopia in Israeli,
Palestinian, Dutch and Belgian populations[14-15,22]. The 2 patients in EQT33 both showed early-onset
symptoms of RP and were found to carry the same p.Lys315* mutation in FAM161A but with different disease severity and
phenotypes. The proband in this family suffered from night blindness, and the
test of visual acuity revealed that both eyes could perceive hand movements,
with minor refraction errors. The OCT showed blurring of the inner and outer
segment layers (IS/OS). The ERG was severely reduced in all six tests. His
visual field was not measured due to his poor visual acuity, while his younger
sister had milder symptoms and signs. Her BCVA was 0.15/0.12 for the right and
left eyes, respectively, with refraction errors of about -10 diopter. Her
fundus showed less pigmentation than that of her brother. Neither patient
suffered from cataracts or other ocular or systemic diseases.
Animal models have proven that the FAM161A protein localizes at the base of the
connecting cilium of photoreceptor cells and is mainly involved in ciliopathy[23-24]. FAM161A is involved in the stabilization of
microtubules, so it is essential for molecular transport from the inner to the
outer segment of the cilium, a function that is critical for the formation of
the outer segment disk[25-26]. Gene-trapped
mice exhibited disorganized discs in their photoreceptor cells and early loss
of photoreceptor function[25]. The mutation c.943A>T identified in pedigree EQT33 introduces a
termination codon, resulting in the translation of a truncated protein, which
severely affects its function. In addition, this mutation is rated as “likely
pathogenic” according to the American College of Medical Genetics and Genomics
(ACMG) standards and guidelines for the interpretation of sequence variants[27].
Segregation analysis revealed co-segregation of the mutation with the disease
phenotype. In summary, we consider the nonsense mutation in FAM161A to be a disease-causing mutation in this
family.
The human RP1L1
gene is on chromosome 8p and consists of 4 exons. It encodes the RP1L1 protein, which is 2400 amino acids long[28]. RP1L1 strongly resembles RP1, mostly within the
first 350 amino acids, including the doublecortin domains[29]. Animal
models have already proven that, like RP1, RP1L1 is specifically expressed in the retina,
especially in the cone and rod photoreceptors, and that it has fundamental
roles in maintaining the photosensitivity and outer segment morphogenesis of
rod photoreceptors[30-31]. Since RP1
has been identified to cause 5.5% and 1% of dominant and recessive RP,
respectively, it is reasonable to deduce that RP1L1 is also a main causative gene of RP. However,
until now, only a few articles have reported a causative relationship between
RP1L1 and RP[32-36]. Most of
the mutations in RP1L1 have been
identified as causing occult macular dysfunction (OMD)[28,31,36-42]. Like most
hereditary ocular diseases, the relationship between RP1L1 genotypes and retinal dystrophy
phenotypes is highly heterogenous. Okuno et al[40] observed a
late and nonsynchronous onset of OMD, while Hayashi et al[42] reported an
early onset of OMD in a Japanese cohort. The mutation frequency for RP among
different ethnicities also differs greatly. Bowne S.J. et al. did not discover
any disease-causing mutations in RP1L1
among 60 AR RP patients in the USA[28],
Haer-Wigman et al[35] detected one mutation causing RP in 266 Dutch
visually impaired patients, and Patel et al[34] revealed
one RP-causing mutation in a Saudi Arabian cohort of 292 families, while
Japanese researchers demonstrated that mutations in RP1L1 are responsible for 7.8% of the AR RP cases in a
Japanese population[36]. In our study, we revealed an AR RP pedigree
harbouring compound heterogeneous mutations (c.56C>A, p.Pro19His; c.5470C>T, p.Gln1824*) in RP1L1, of which the missense mutation (c.56C>A, p.Pro19His) is predicted to be
probably damaging, to be deleterious and to have a damaging impact on protein
function by Poly-Phen2, PROVEN and SIFT, respectively; the nonsense mutation
(p.Gln1824*) leads to the expression of a truncated protein or, more likely,
results in nonsense-mediated decay. As in to the report by Okuno et al[40], the fundus
phenotype of one proband was asymmetrical, but since she was only 26 years old,
we speculate that the second eye may exhibit signs of RP in the future,
otherwise there is occurrence of de novel mutation or germline mosaic. This
proband came from Southwest China, which is an extremely impoverished region.
It is unfortunate that this proband temporarily decided not to participate in
follow-up, but we will not cease tracing the advance of the disease or
verifying the unique phenotype exhibited by this family.
In summary, we identified 2 novel mutations in genes
responsible for AR RP, and the mutation in FAM161A is reported for the first time in a Chinese
population to cause AR RP. Due to limited data about the RP1L1 mutation, more studies are required to
provide more evidence regarding the role of this mutation. Nevertheless, our
study enriches the knowledge of the mutation frequency and spectrum in the
genes responsible for RP and provides a new target for future gene therapy.
ACKNOWLEDGEMENTS
We sincerely thank all the subjects participated in this
study.
Foundation: Supported by the National
Natural Science Foundation of China (No.81360154).
Conflicts of Interest: Hu YS, None;
Song H, None; Li Y, None; Xiao ZY, None; Li T, None.
REFERENCES
|
1 Narayan DS, Wood JP,
Chidlow G, Casson RJ. A review of the mechanisms of cone degeneration in
retinitis pigmentosa. Acta Ophthalmol 2016;94(8):748-754.
https://doi.org/10.1111/aos.13141
PMid:27350263
|
|
|
|
2 Zobor D, Zrenner E.
Retinitis pigmentosa - a review. Pathogenesis, guidelines for diagnostics and
perspectives. Ophthalmologe 2012;109(5): 501-514;quiz 515.
https://doi.org/10.1007/s00347-012-2555-6
PMid:22581051
|
|
|
|
|
3 Ali MU, Rahman MSU,
Cao J, Yuan PX. Genetic characterization and disease mechanism of retinitis
pigmentosa; current scenario. 3 Biotech 2017;7(4):251.
https://doi.org/10.1007/s13205-017-0878-3
PMid:28721681 PMCid:PMC5515732
|
|
|
|
|
4 Hartong DT, Berson
EL, Dryja TP. Retinitis pigmentosa. Lancet 2006;368(9549):1795-1809.
https://doi.org/10.1016/S0140-6736(06)69740-7
|
|
|
|
|
5 Hu DN. Prevalence
and mode of inheritance of major genetic eye diseases in China. J Med Genet
1987;24(10):584-588.
https://doi.org/10.1136/jmg.24.10.584
PMid:3500313 PMCid:PMC1050283
|
|
|
|
|
6 Xu Y, Guan L, Shen
T, Zhang J, Xiao X, Jiang H, Li S, Yang J, Jia X, Yin Y, Guo X, Wang J, Zhang
Q. Mutations of 60 known causative genes in 157 families with retinitis
pigmentosa based on exome sequencing. Hum Genet 2014;133(10):1255-1271.
https://doi.org/10.1007/s00439-014-1460-2
PMid:24938718
|
|
|
|
|
7 Beryozkin A, Shevah
E, Kimchi A, Mizrahi-Meissonnier L, Khateb S, Ratnapriya R, Lazar CH,
Blumenfeld A, Ben-Yosef T, Hemo Y, Pe'er J, Averbuch E, Sagi M, Boleda A,
Gieser L, Zlotogorski A, Falik-Zaccai T, Alimi-Kasem O, Jacobson SG, Chowers
I, Swaroop A, Banin E, Sharon D. Whole exome sequencing reveals mutations in
known retinal disease genes in 33 out of 68 israeli families with inherited
retinopathies. Sci Rep 2015;5:13187.
https://doi.org/10.1038/srep13187
PMid:26306921 PMCid:PMC4549705
|
|
|
|
|
8 Huang L, Zhang Q,
Huang X, Qu C, Ma S, Mao Y, Yang J, Li Y, Li Y, Tan C, Zhao P, Yang Z.
Mutation screening in genes known to be responsible for retinitis pigmentosa
in 98 Small Han Chinese Families. Sci Rep 2017;7(1):1948.
https://doi.org/10.1038/s41598-017-00963-6
PMid:28512305 PMCid:PMC5434011
|
|
|
|
|
9 Xin W, Xiao X, Li
SQ, Zhang Q. Late-onset CORD in a patient with RDH12 mutations identified by
whole exome sequencing. Ophthalmic Genet 2016;37(3):345-348.
https://doi.org/10.3109/13816810.2015.1059457
PMid:26848971
|
|
|
|
|
10 Zhao F,
Wu J, Xue A, Su Y, Wang X, Lu X, Zhou Z, Qu J, Zhou X. Exome sequencing
reveals CCDC111 mutation associated with high myopia. Hum Genet
2013;132(8):913-921.
https://doi.org/10.1007/s00439-013-1303-6
PMid:23579484
|
|
|
|
|
11
McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, Bach M.
Erratum to: ISCEV Standard for full-field clinical electroretinography (2015 update).
Doc Ophthalmol 2015;131(1):81-83.
https://doi.org/10.1007/s10633-015-9504-z
PMid:26059396
|
|
|
|
|
12
Langmann T, Di Gioia SA, Rau I, Stöhr H, Maksimovic NS, Corbo JC, Renner AB,
Zrenner E, Kumaramanickavel G, Karlstetter M, Arsenijevic Y, Weber BH, Gal A,
Rivolta C. Nonsense mutations in FAM161A cause RP28-associated recessive
retinitis pigmentosa. Am J Hum Genet 2010;87(3):376-381.
https://doi.org/10.1016/j.ajhg.2010.07.018
PMid:20705278 PMCid:PMC2933350
|
|
|
|
|
13
Venturini G, Di Gioia SA, Harper S, Weigel-DiFranco C, Rivolta C, Berson EL. Molecular
genetics of FAM161A in North American patients with early-onset retinitis
pigmentosa. PLoS One 2014;9(3):e92479.
https://doi.org/10.1371/journal.pone.0092479
PMid:24651477 PMCid:PMC3961368
|
|
|
|
|
14 Van
Schil K, Klevering BJ, Leroy BP, Pott JW, Bandah-Rozenfeld D,
Zonneveld-Vrieling MN, Sharon D, den Hollander AI, Cremers FP, De Baere E,
Collin RW, van den Born LI. A nonsense mutation in FAM161A is a recurrent
founder allele in dutch and belgian individuals with autosomal recessive
retinitis pigmentosa. Invest Ophthalmol Vis Sci 2015;56(12):7418-7426.
https://doi.org/10.1167/iovs.15-17920
PMid:26574802
|
|
|
|
|
15
Bandah-Rozenfeld D, Mizrahi-Meissonnier L, Farhy C, Obolensky A, Chowers I,
Pe'er J, Merin S, Ben-Yosef T, Ashery-Padan R, Banin E, Sharon D.
Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive
retinitis pigmentosa. Am J Hum Genet 2010;87(3):382-391.
https://doi.org/10.1016/j.ajhg.2010.07.022
PMid:20705279 PMCid:PMC2933343
|
|
|
|
|
16
O'Sullivan J, Mullaney BG, Bhaskar SS, Dickerson JE, Hall G, O'Grady A,
Webster A, Ramsden SC, Black GC. A paradigm shift in the delivery of services
for diagnosis of inherited retinal disease. J Med Genet 2012;49(5):322-326.
https://doi.org/10.1136/jmedgenet-2012-100847
PMid:22581970
|
|
|
|
|
17
Carmichael H, Shen Y, Nguyen TT, Hirschhorn JN, Dauber A. Whole exome
sequencing in a patient with uniparental disomy of chromosome 2 and a complex
phenotype. Clin Genet 2013;84(3):213-222.
https://doi.org/10.1111/cge.12064
PMid:23167750 PMCid:PMC3996682
|
|
|
|
|
18 Maranhao
B, Biswas P, Gottsch AD, Navani M, Naeem MA, Suk J, Chu J, Khan SN, Poleman
R, Akram J, Riazuddin S, Lee P, Riazuddin SA, Hejtmancik JF, Ayyagari R.
Investigating the molecular basis of retinal degeneration in a familial
cohort of pakistani decent by exome sequencing. PLoS One 2015;10(9):e0136561.
https://doi.org/10.1371/journal.pone.0136561
PMid:26352687 PMCid:PMC4564165
|
|
|
|
|
19 Rose AM,
Sergouniotis P, Alfano G, Muspratt-Tucker N, Barton S, Moore AT, Black G,
Bhattacharya SS, Webster AR. Diverse clinical phenotypes associated with a
nonsense mutation in FAM161A. Eye (Lond) 2015;29(9):1226-1232.
https://doi.org/10.1038/eye.2015.93
PMid:26113502 PMCid:PMC4565954
|
|
|
|
|
20 Zhou Y,
Saikia BB, Jiang Z, Zhu X, Liu Y, Huang L, Kim R, Yang Y, Qu C, Hao F, Gong
B, Tai Z, Niu L, Yang Z, Sundaresan P, Zhu X. Whole-exome sequencing reveals
a novel frameshift mutation in the FAM161A gene causing autosomal recessive
retinitis pigmentosa in the Indian population. J Hum Genet
2015;60(10):625-630.
https://doi.org/10.1038/jhg.2015.92
PMid:26246154
|
|
|
|
|
21 Duncan
JL, Biswas P, Kozak I, Navani M, Syed R, Soudry S, Menghini M, Caruso RC,
Jeffrey BG, Heckenlively JR, Reddy GB, Lee P, Roorda A, Ayyagari R. Ocular phenotype
of a family with FAM161A-associated retinal degeneration. Ophthalmic Genet
2016;37(1):44-52.
https://doi.org/10.3109/13816810.2014.929716
PMid:25007332 PMCid:PMC4289132
|
|
|
|
|
22 Zobor
D, Balousha G, Baumann B, Wissinger B. Homozygosity mapping reveals new
nonsense mutation in the FAM161A gene causing autosomal recessive retinitis
pigmentosa in a Palestinian family. Mol Vis 2014;20:178-182.
|
|
|
|
|
23 Zach F,
Stöhr H. FAM161A, a novel centrosomal-ciliary protein implicated in autosomal
recessive retinitis pigmentosa. Adv Exp Med Biol 2014;801:185-190.
https://doi.org/10.1007/978-1-4614-3209-8_24
PMid:24664697
|
|
|
|
|
24 Di
Gioia SA, Letteboer SJ, Kostic C, Bandah-Rozenfeld D, Hetterschijt L, Sharon
D, Arsenijevic Y, Roepman R, Rivolta C. FAM161A, associated with retinitis
pigmentosa, is a component of the cilia-basal body complex and interacts with
proteins involved in ciliopathies. Hum Mol Genet 2012;21(23):5174-5184.
https://doi.org/10.1093/hmg/dds368
PMid:22940612
|
|
|
|
|
25
Karlstetter M, Sorusch N, Caramoy A, Dannhausen K, Aslanidis A, Fauser S,
Boesl MR, Nagel-Wolfrum K, Tamm ER, Jägle H, Stoehr H, Wolfrum U, Langmann T.
Disruption of the retinitis pigmentosa 28 gene Fam161a in mice affects
photoreceptor ciliary structure and leads to progressive retinal
degeneration. Hum Mol Genet 2014;23(19):5197-5210.
https://doi.org/10.1093/hmg/ddu242
PMid:24833722
|
|
|
|
|
26 Zach F,
Grassmann F, Langmann T, Sorusch N, Wolfrum U, Stöhr H. The retinitis pigmentosa
28 protein FAM161A is a novel ciliary protein involved in intermolecular
protein interaction and microtubule association. Hum Mol Genet
2012;21(21):4573-4586.
https://doi.org/10.1093/hmg/dds268
PMid:22791751
|
|
|
|
|
27
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde
M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance
Committee. Standards and guidelines for the interpretation of sequence
variants: a joint consensus recommendation of the American College of Medical
Genetics and Genomics and the Association for Molecular Pathology. Genet Med
2015;17(5):405-424.
https://doi.org/10.1038/gim.2015.30
PMid:25741868 PMCid:PMC4544753
|
|
|
|
|
28 Bowne
SJ, Daiger SP, Malone KA, Heckenlively JR, Kennan A, Humphries P, Hughbanks-Wheaton
D, Birch DG, Liu Q, Pierce EA, Zuo J, Huang Q, Donovan DD, Sullivan LS.
Characterization of RP1L1, a highly polymorphic paralog of the retinitis
pigmentosa 1 (RP1) gene. Mol Vis 2003;9:129-137.
|
|
|
|
|
29 Conte
I, Lestingi M, den Hollander A, Alfano G, Ziviello C, Pugliese M, Circolo D,
Caccioppoli C, Ciccodicola A, Banfi S. Identification and characterisation of
the retinitis pigmentosa 1-like1 gene (RP1L1): a novel candidate for retinal
degenerations. Eur J Hum Genet 2003;11(2):155-162.
https://doi.org/10.1038/sj.ejhg.5200942
PMid:12634863
|
|
|
|
|
30
Yamashita T, Liu J, Gao J, LeNoue S, Wang C, Kaminoh J, Bowne SJ, Sullivan LS,
Daiger SP, Zhang K, Fitzgerald ME, Kefalov VJ, Zuo J. Essential and
synergistic roles of RP1 and RP1L1 in rod photoreceptor axoneme and retinitis
pigmentosa. J Neurosci 2009;29(31):9748-9760.
https://doi.org/10.1523/JNEUROSCI.5854-08.2009
PMid:19657028 PMCid:PMC2748320
|
|
|
|
|
31 Akahori
M, Tsunoda K, Miyake Y, Fukuda Y, Ishiura H, Tsuji S, Usui T, Hatase T,
Nakamura M, Ohde H, Itabashi T, Okamoto H, Takada Y, Iwata T. Dominant
mutations in RP1L1 are responsible for occult macular dystrophy. Am J Hum
Genet 2010;87(3):424-429.
https://doi.org/10.1016/j.ajhg.2010.08.009
PMid:20826268 PMCid:PMC2933341
|
|
|
|
|
32
Davidson AE, Sergouniotis PI, Mackay DS, Wright GA, Waseem NH, Michaelides M,
Holder GE, Robson AG, Moore AT, Plagnol V, Webster AR. RP1L1 variants are
associated with a spectrum of inherited retinal diseases including retinitis
pigmentosa and occult macular dystrophy. Hum Mutat 2013;34(3):506-514.
https://doi.org/10.1002/humu.22264
PMid:23281133
|
|
|
|
|
33 Liu YP,
Bosch DG, Siemiatkowska AM, Rendtorff ND, Boonstra FN, Möller C, Tranebjærg
L, Katsanis N, Cremers FP. Putative digenic inheritance of heterozygous RP1L1
and C2orf71 null mutations in syndromic retinal dystrophy. Ophthalmic Genet
2017;38(2):127-132.
https://doi.org/10.3109/13816810.2016.1151898
PMid:27029556 PMCid:PMC5967407
|
|
|
|
|
34 Patel
N, Aldahmesh MA, Alkuraya H, et al. Expanding the clinical, allelic, and
locus heterogeneity of retinal dystrophies. Genet Med 2016;18(6):554-562.
https://doi.org/10.1038/gim.2015.127
PMid:26355662
|
|
|
|
|
35
Haer-Wigman L, van Zelst-Stams WA, Pfundt R, et al. Diagnostic exome
sequencing in 266 Dutch patients with visual impairment. Eur J Hum Genet
2017;25(5):591-599.
https://doi.org/10.1038/ejhg.2017.9
PMid:28224992 PMCid:PMC5437915
|
|
|
|
|
36 Oishi M,
Oishi A, Gotoh N, Ogino K, Higasa K, Iida K, Makiyama Y, Morooka S, Matsuda
F, Yoshimura N. Comprehensive molecular diagnosis of a large cohort of
Japanese retinitis pigmentosa and Usher syndrome patients by next-generation
sequencing. Invest Ophthalmol Vis Sci 2014;55(11):7369-7375.
https://doi.org/10.1167/iovs.14-15458
PMid:25324289
|
|
|
|
|
37 Kato Y,
Hanazono G, Fujinami K, Hatase T, Kawamura Y, Iwata T, Miyake Y, Tsunoda K.
Parafoveal photoreceptor abnormalities in asymptomatic patients with RP1L1
mutations in families with occult macular dystrophy. Invest Ophthalmol Vis
Sci 2017;58(14):6020-6029.
https://doi.org/10.1167/iovs.17-21969
PMid:29196766
|
|
|
|
|
38
Piermarocchi S, Segato T, Leon A, Colavito D, Miotto S. Occult macular
dystrophy in an Italian family carrying a mutation in the RP1L1 gene. Mol Med
Rep 2016;13(3):2308-2312.
https://doi.org/10.3892/mmr.2016.4784
PMid:26782618
|
|
|
|
|
39
Fujinami K, Kameya S, Kikuchi S, et al. Novel RP1L1 variants and
genotype-photoreceptor microstructural phenotype associations in cohort of
japanese patients with occult macular dystrophy. Invest Ophthalmol Vis Sci
2016;57(11):4837-4846.
https://doi.org/10.1167/iovs.16-19670
PMid:27623337
|
|
|
|
|
40 Okuno T,
Hayashi T, Sugasawa J, Oku H, Yamada H, Tsuneoka H, Ikeda T. Elderly case of
pseudo-unilateral occult macular dystrophy with Arg45Trp mutation in RP1L1
gene. Doc Ophthalmol 2013;127(2):141-146.
https://doi.org/10.1007/s10633-013-9384-z
PMid:23619761
|
|
|
|
|
41 Tsunoda
K, Usui T, Hatase T, Yamai S, Fujinami K, Hanazono G, Shinoda K, Ohde H,
Akahori M, Iwata T, Miyake Y. Clinical characteristics of occult macular
dystrophy in family with mutation of RP1l1 gene. Retina 2012;32(6):1135-1147.
https://doi.org/10.1097/IAE.0b013e318232c32e
PMid:22466457
|
|
|
|
|
42 Hayashi
T, Gekka T, Kozaki K, Ohkuma Y, Tanaka I, Yamada H, Tsuneoka H. Autosomal
dominant occult macular dystrophy with an RP1L1 mutation (R45W). Optom Vis
Sci 2012;89(5):684-691.
https://doi.org/10.1097/OPX.0b013e31824eea32
PMid:22504327
|
|
|
|
Citation:
Hu YS, Song H, Li Y, Xiao ZY, Li T. Whole-exome sequencing identifies novel mutations
in genes responsible for retinitis pigmentosa in 2 nonconsanguineous Chinese
families. Int J Ophthalmol
2019;12(6):915-923
DOI:10.18240/ijo.2019.06.06