
DOI:10.18240/ijo.2019.07.01
Citation: Zhou
FQ, Wang QW, Liu ZZ, Zhang XL, Wang DN, Dongye MM, Lin HT, Chen WR. Novel
mutation in OCRL leading to a severe form of Lowe syndrome. Int J
Ophthalmol 2019;12(7):1057-1060
・Basic Research・
Novel
mutation in OCRL leading to a severe form of Lowe syndrome
Feng-Qi Zhou1,2, Qi-Wei Wang1,
Zhen-Zhen Liu1, Xu-Lin Zhang1, Dong-Ni Wang1,
Mei-Mei Dongye1, Hao-Tian Lin1, Wei-Rong Chen1
1State Key
Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen
University, Guangzhou 510060, Guangdong Province, China
2New England
College of Optometry, Boston, MA 02115, USA
Co-first
authors: Feng-Qi Zhou
and Qi-Wei Wang
Correspondence
to: Wei-Rong
Chen and Hao-Tian Lin. State Key Laboratory of Ophthalmology, Zhongshan
Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, Guangdong
Province, China. chenwr_q@aliyun.com; haot.lin@hotmail.com
Received:
2018-10-16 Accepted:
2019-02-01
Abstract
AIM: To
investigate the phenotype and genotype of a family with X-linked recessive Lowe
syndrome.
METHODS: All the
members in the Chinese pedigree underwent comprehensive ophthalmologic and
systemic examinations. Genomic DNA was isolated from peripheral blood of the
pedigree members and 100 unrelated healthy Chinese subjects. Direct sequencing
was performed to screen the exons and intron boundaries of OCRL.
RESULTS: The
ophthalmological and systemic examinations suggested that the affected
individual had Lowe syndrome. The phenotype in the pedigree is severe and
consistent among all the affected individuals except for an individual who
additionally suffered from congenital heart disease and laryngeal cartilage
dysplasia. Directional Sanger sequencing identified a complex mutation c.(2368_2368delG;
c
.2370A>C)
in the Rho-GTPase activating protein domain. This complex mutation causes
termination of protein synthesis at amino acid 824 and result in a new peptide
with 823 amino acids (p.Ala790ProfsX34). This mutation was not detected in 100
unrelated healthy Chinese subjects.
CONCLUSION: Our
findings expand the phenotypic and genotypic spectrum of Lowe syndrome.
KEYWORDS: Lowe
syndrome; oculocerebrorenal syndrome; OCRL; congenital membranous
cataract
DOI:10.18240/ijo.2019.07.01
Citation: Zhou
FQ, Wang QW, Liu ZZ, Zhang XL, Wang DN, Dongye MM, Lin HT, Chen WR. Novel
mutation in OCRL leading to a severe form of Lowe syndrome. Int J
Ophthalmol 2019;12(7):1057-1060
INTRODUCTION
Lowe
oculocerebrorenal syndrome[1] (OMIM #309000; Lowe
syndrome) is a very rare X-linked recessive pathology which is characterized by
multiple disorders involving the eyes, the central nervous system, and the
kidneys[2]. The prevalence of this disease ranges
between 1:500 000 to 1:1 000 000[3]. The causative
gene OCRL, located on chromosome Xq26.1, encodes an inositol
polyphosphate 5-phosphatase[4-6].
There are approximately 250 mutations of OCRL gene reported causing Lowe
syndrome according to the Human Gene Mutation Database (HGMD)[7].
The most commonly mutated category was missense and nonsense mutations (49%),
followed by small deletions (20%), splicing mutations (12%), small insertions
(9%), complete gene deletion, and large insertion.
Herein, we
present a Lowe syndrome pedigree with a complex mutation in OCRL gene. We
expand the phenotype and genotype of Lowe syndrome.
SUBJECTS AND METHODS
Ethical
Approval Informed written consent was
obtained from each participant or legal guardian according to the tenets of the
Declaration of Helsinki. The research protocol was approved by the
Institutional Review Board/Ethics Committee of Sun Yat-sen University
(Guangzhou, China).
A family
with Lowe syndrome was recruited in the Zhongshan Ophthalmic Center, Sun
Yat-sen University, Guangzhou, China. Complete disease history was taken.
Comprehensive ophthalmic and systemic examinations were performed in the
proband and his family members. Eight individuals from the family participated
in the present study (only disease history was available for the three deceased
individuals). One hundred unrelated Chinese subjects were recruited as
controls. Genomic DNA was isolated from peripheral blood following
manufacture’s instruction (TIANGEN Biotech Co. Ltd., Beijing, China). Exons and
intron boundaries of OCRL were screened by direct sequencing. The
polymerase chain reaction (PCR) was carried out with condition as previously
described[8]. The PCR products were sequenced
using the BigDye Terminator Cycle sequencing kit (ABI Applied Biosystems;
Sangon Co., Shanghai, China).
RESULTS
We identified
a family with X-linked congenital cataract (Figure
1A). The proband was an 8-month-old boy (III:6, Figure
1B) born to non-consanguineous Chinese parents. He was delivered after a
full-term pregnancy. Muscular hypotonia, hypoxic-ischemic encephalopathy, and
undescended testicle were diagnosed at birth, and congenital cataract was
diagnosed at the age of 1mo. His development was markedly delayed. At the age
of 8mo, he still could not raise his head, turn over and sit by himself. Apart
from membranous cataract, a severe form of congenital cataract, the proband
presented with small pupils which were difficult to be dilated. The largest
pupil diameter was
5 mm in the
right eye and
3.5 mm in the
left eye after mydriasis using compound tropicamide. Abnormality in urine
protein test (urine protein ++) and blood coagulation (thrombin time 22.9s)
were noticed as a preoperative check before cataract extraction. Further
examination detected a renal tubular dysfunction (beta-microglobulin 77.20
mg/L, beta-microglobulin/creatinine 63591.43 μg/mmol, retinol-binding protein
3.26 mg/L, alpha-microglobulin 110.0 mg/L, N-acetyl-β-D-glucosaminidase 30.7
U/L).
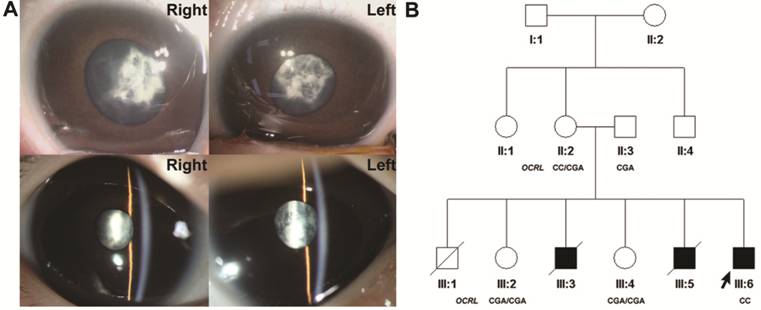
Figure 1
Slit-lamp photographs of the proband and family pedigree A: Slit-lamp photographs of the
proband of the pedigree illustrating membranous cataracts; B: Pedigree drawing
with the genotypes of OCRL. The proband is marked with an arrow. Square
and circle indicate male and female respectively. Filled and blank symbols
represent affected and unaffected individuals, respectively. Diagonal line
through a symbol represent deceased individual.
The parents
(II:2 and II:3) and the sisters (III:2 and III:4) of the proband are healthy
but all the brothers of the proband died in early age. The first child (III:1)
of the family was a boy, and he was delivered after a full-term pregnancy. He
died during delivery for unknown reason. Therefore, the diagnosis of Lowe is
undetermined. The second boy (III:3) in the pedigree died of hypoxic-ischemic
encephalopathy at 1.5-month old. He also suffered from congenital cataract and
undescended testicle. The third boy (III:5) of this family died during the
surgery of congenital heart disease at the age of 8mo. He also suffered from
congenital cataract, hypoxic-ischemic encephalopathy, laryngeal cartilage dysplasia,
undescended testicle, and developmental delay. No abnormality was detected in
the hearing test of all the children in the pedigree. The ophthalmological and
systemic evaluations, as well as the medical history of the family, were
consistent with a diagnosis of Lowe syndrome.
By direct
sequencing of the coding and flanking regions of OCRL (NM_000276.3, MIM
number 300535), a complex mutation c.(2368_2368delG; c
.2370A>C) was detected in the proband (III:6). The
mutation (Figure
2A) is
located in exon 21 of OCRL. Deletion of a single base pair at nucleotide
position 2368 (c.2368_2368delG) and a variant at nucleotide position 2370 (c
.2370A>C) were located in
the same allele causing the termination of protein synthesis at amino acid 824
and result in a new peptide with 823 amino acids (p.Ala790ProfsX34). The
heterozygous variants were detected in the patient’s mother. Both variants have
not been detected in the patient’s father (II:3), the patient’s sisters (III:2
and III:4), the 100 healthy Chinese adults, the ExAC database, and the 1000
Genomes database. No previously report was found in the literature.
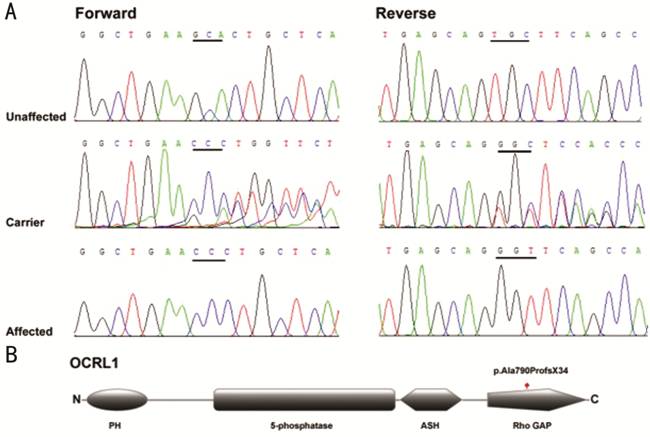
Figure 2
Genetic information A: Sequence chromatograms of the
c.2368_2368delG and c
.2370A>C
(p.Ala790ProfsX34) variation identified; B: Domain structure of OCRL1 with the
complex mutation. Graphic overview of the protein encoded by OCRL.
Structural or functional domains are depicted, as well as the position of the
mutation. PH: Pleckstrin homology domain; 5-phosphatase: 5-phosphatase
catalytic domain; ASH: ASPM, SPD-2, Hydin domain; Rho GAP: Rho-GAPase
activating protein domain.
DISCUSSION
In the
present study, we detected a complex mutation c.(2368_2368delG; c
.2370A>C) in one allele of OCRL in a Chinese family with Lowe syndrome. In this family, all the sons
died in a very early age without sequence data except for the proband. Both
daughters are healthy without any sign of Lowe syndrome. Although previous
studies suggested the mutation in OCRL showed phenotypic heterogeneity[9-11], the present pedigree had relatively
similar phenotype among the affected and the suspected affected family members.
III:6, III:3, and III:5 showed severe hypoxic-ischemic encephalopathy, severe
muscular hypotonia, bilateral congenital cataract, and undescended testicle.
Notably, III:5 was additionally diagnosed with congenital heart disease and
laryngeal dysplasia.
Mutations in OCRL gene, encoding inositol polyphosphate 5-phosphatase, have been
reported causing the Lowe syndrome. The mRNA transcript of full-length OCRL gene contains 24 exons, including an alternatively spliced
18a exon. Therefore, two isoforms are
produced. Isoform A (OCRL1 protein) encodes 8 more amino acids than isoform B,
and is expressed ubiquitously. In contrast, isoform B is not expressed in the
brain[4,12]. Most OCRL mutations associated with Lowe syndrome are located in exons 8 to 23[3,7,13]. OCLR1
contains 4 domains (Figure 2B). The N-terminal pleckstrin homology (PH) domain
(encoded by exon 2-5) plays a part in recognizing phosphatidylinositol 4,5‑bisphosphate
[PI(4,5)P2] on the membrane[14]. The
central 5-phosphatase catalytic domain (encoded by exon 9-15) shows catalytic
properties[15-16]. The ASH
(ASPM, SPD-2, Hydin) domain and the C-terminal noncatalytic Rho-GTPase
activating protein (Rho GAP) domain (encoded by exon 16-22) mediate the
interaction of OCRL1 and its partners[17].
Lowe
syndrome is caused by partial or complete loss of PI(4,5)P2ase
activity[18-19]. The missense
mutations are likely to produce less OCRL1 protein with decreased PI(4,5)P2ase
activity[10]. The possible mechanism of the
decrease amount of the OCRL1 with missense mutation is that the deleterious
OCRL1 induced the activation of the endoplasmic reticulum-associated
degradation or of the unfolded protein response[20].
For nonsense, frameshift, deletion, and splicing mutations, the mutations were
associated with low mRNA content which was likely to be caused by the decreased
transcription and nonsense mediated decay[7,10,21-22]. In the
present pedigree, we identified a complex mutation. Although the c
.2370A>C is a synonymous
mutation, there was a deletion (c.2368_2368delG) occurred two base pairs ahead
of it. The combined effect of the two variants is the generation of a shorter
new peptide (823 amino acids) than usual, and the amino acid 790 changes from
alanine (reference) and histidine (c.2368_2368delG only) to proline
(c.2368_2368delG and c
.2370A>C).
The pathogenic mechanism involved in this complex mutation is probably nonsense
mediated decay of the mRNA induced by the premature stop codon. Moreover, the
present mutation resulting in shorter protein with incomplete Rho GAP domain
might have an impact on the interaction between OCRL1 and its partners.
However, further study is needed to elucidate the actual pathogenic mechanism.
The types of
mutations are unlikely to correlate with the severity of the disease. Even in
the same family, the phenotypes of the affected individuals showed
heterogeneity. The variability was explained by the difference in the patients’
genetic background[9-10,23-24]. In contrast, the phenotype of the present family
showed intrafamiliar consistency. The three members (III:3, III:5, and III:6)
shared similar phenotype (bilateral congenital cataract, severe
hypoxic-ischemic encephalopathy, severe muscular hypotonia, and undescended
testicle). However, III:5 suffered from congenital heart disease and laryngeal
dysplasia which might indicate some inconsistency. Apart from genetic
background difference, a potential explanation is that the patients (III:3 and
III:6) were still young and hasn’t shown the symptom yet.
Additionally,
bilateral pupils difficult to be dilated, congenital heart disease, and
laryngeal dysplasia are the symptom which were not reported in previous
studies. Since OCRL expresses ubiquitously, including eye and heart, the
occurrence of abnormality in iris and heart suggests Lowe syndrome might also
have an impact on the uvea and cardiovascular system. This correlation needs to
be validated by more Lowe syndrome pedigrees.
In summary,
we identified a complex mutation c.(2368_2368delG; c
.2370A>C) of OCRL in a Chinese family. The
phenotype in this pedigree includes severe hypoxic-ischemic encephalopathy,
severe muscular hypotonia, congenital heart disease, laryngeal dysplasia,
bilateral congenital membranous cataracts, bilateral pupils difficult to be
dilated, and undescended testicle. Our findings expand the phenotypic and
genotypic spectrum of Lowe syndrome.
ACKNOWLEDGEMENTS
The authors
are grateful to all members in the family for their participation in the study.
Foundations: Supported by the National Natural
Science Foundation of China (No.81700812); the Ph.D. Start-up Fund of Natural
Science Foundation of Guangdong Province (No
.2017A030310214); the Guangdong Provincial Foundation
for Medical Scientific Research (No.A2017016).
Conflicts of
Interest: Zhou FQ, None; Wang QW, None; Liu ZZ, None; Zhang XL, None; Wang
DN, None; Dongye MM, None; Lin HT, None; Chen WR, None.
REFERENCES
1 Lowe CU, Terrey M, MacLACHLAN EA.
Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and
mental retardation; a clinical entity. AMA Am J Dis Child 1952;83(2):164-184.
https://doi.org/10.1001/archpedi.1952.02040060030004 |
| |
2 Bökenkamp A, Ludwig M. The
oculocerebrorenal syndrome of Lowe: an update. Pediatr Nephrol
2016;31(12):2201-2212.
https://doi.org/10.1007/s00467-016-3343-3
PMid:27011217 PMCid:PMC5118406 |
|
| |
3 De Matteis MA, Staiano L, Emma F, Devuyst
O. The 5-phosphatase OCRL in Lowe syndrome and Dent disease 2. Nat Rev
Nephrol 2017;13(8):455-470.
https://doi.org/10.1038/nrneph.2017.83
PMid:28669993 |
|
| |
4 Attree O, Olivos IM, Okabe I, Bailey LC,
Nelson DL, Lewis RA, McInnes RR, Nussbaum RL. The Lowe's oculocerebrorenal
syndrome gene encodes a protein highly homologous to inositol
polyphosphate-5-phosphatase. Nature 1992;358(6383):239-242.
https://doi.org/10.1038/358239a0
PMid:1321346 |
|
| |
5 Suchy SF, Olivos-Glander IM, Nussabaum
RL. Lowe syndrome, a deficiency of phosphatidylinositol 4, 5-bisphosphate
5-phosphatase in the Golgi apparatus. Hum Mol Genet 1995;4(12):2245-2250.
https://doi.org/10.1093/hmg/4.12.2245
PMid:8634694 |
|
| |
6 Zhang X, Jefferson AB, Auethavekiat V,
Majerus PW. The protein deficient in Lowe syndrome is a
phosphatidylinositol-4, 5-bisphosphate 5-phosphatase. Proc Natl Acad Sci U S
A 1995;92(11):4853-4856.
https://doi.org/10.1073/pnas.92.11.4853
PMid:7761412 PMCid:PMC41805 |
|
| |
7 Suarez-Artiles L, Perdomo-Ramirez A,
Ramos-Trujillo E, Claverie-Martin F. Splicing analysis of exonic OCRL
mutations causing lowe syndrome or dent-2 disease. Genes (Basel)
2018;9(1):E15.
https://doi.org/10.3390/genes9010015
PMid:29300302 PMCid:PMC5793168 |
|
| |
8 Jin CF, Wang QW, Li JN, Zhu YN, Shentu
XC, Yao K. A recurrent PAX6 mutation is associated with aniridia and
congenital progressive cataract in a Chinese family. Mol Vis 2012;18:465-470. |
|
| |
9 Zaniew M, Bökenkamp A, Kolbuc M, et al.
Long-term renal outcome in children with OCRL mutations: retrospective
analysis of a large international cohort. Nephrol Dial Transplant
2018;33(1):85-94. |
|
| |
10 Hichri H, Rendu J, Monnier
N, Coutton C, Dorseuil O, Poussou RV, Baujat G, Blanchard A, Nobili F,
Ranchin B, Remesy M, Salomon R, Satre V, Lunardi J. From Lowe syndrome to
Dent disease: correlations between mutations of the OCRL1 gene and clinical
and biochemical phenotypes. Hum Mutat 2011;32(4):379-388.
https://doi.org/10.1002/humu.21391
PMid:21031565 |
|
| |
11 Montjean R, Aoidi R, Desbois
P, Rucci J, Trichet M, Salomon R, Rendu J, Fauré J, Lunardi J, Gacon G,
Billuart P, Dorseuil O. OCRL-mutated fibroblasts from patients with Dent-2
disease exhibit INPP5B-independent phenotypic variability relatively to Lowe
syndrome cells. Hum Mol Genet 2015;24(4):994-1006.
https://doi.org/10.1093/hmg/ddu514
PMid:25305077 |
|
| |
12 Johnson JM, Castle J,
Garrett-Engele P, Kan ZY, Loerch PM, Armour CD, Santos R, Schadt EE,
Stoughton R, Shoemaker DD. Genome-wide survey of human alternative pre-mRNA
splicing with exon junction microarrays. Science 2003;302(5653):2141-2144.
https://doi.org/10.1126/science.1090100
PMid:14684825 |
|
| |
13 Charnas LR, Bernardini I,
Rader D, Hoeg JM, Gahl WA. Clinical and laboratory findings in the oculocerebrorenal
syndrome of Lowe, with special reference to growth and renal function. N Engl
J Med 1991;324(19):1318-1325.
https://doi.org/10.1056/NEJM199105093241904
PMid:2017228 |
|
| |
14 Noakes CJ, Lee G, Lowe M.
The PH domain proteins IPIP27A and B link OCRL1 to receptor recycling in the
endocytic pathway. Mol Biol Cell 2011;22(5):606-623.
https://doi.org/10.1091/mbc.e10-08-0730
PMid:21233288 PMCid:PMC3046058 |
|
| |
15 Whisstock JC, Wiradjaja F,
Waters JE, Gurung R. The structure and function of catalytic domains within
inositol polyphosphate 5-phosphatases. IUBMB Life 2002;53(1):15-23.
https://doi.org/10.1080/15216540210814
PMid:12018403 |
|
| |
16 Pirruccello M, De Camilli P.
Inositol 5-phosphatases: insights from the Lowe syndrome protein OCRL. Trends
Biochem Sci 2012;37(4): 134-143.
https://doi.org/10.1016/j.tibs.2012.01.002
PMid:22381590 PMCid:PMC3323734 |
|
| |
17 McCrea HJ, Paradise S,
Tomasini L, Addis M, Melis MA, De Matteis MA, De Camilli P. All known patient
mutations in the ASH-RhoGAP domains of OCRL affect targeting and APPL1
binding. Biochem Biophys Res Commun 2008;369(2):493-499.
https://doi.org/10.1016/j.bbrc.2008.02.067
PMid:18307981 PMCid:PMC2442618 |
|
| |
18 Tsujishita Y, Guo S, Stolz
LE, York JD, Hurley JH. Specificity determinants in phosphoinositide
dephosphorylation: crystal structure of an archetypal inositol polyphosphate
5-phosphatase. Cell 2001;105(3):379-389.
https://doi.org/10.1016/S0092-8674(01)00326-9 |
|
| |
19 De Leo MG, Staiano L,
Vicinanza M, et al. Autophagosome-lysosome fusion triggers a lysosomal
response mediated by TLR9 and controlled by OCRL. Nat Cell Biol
2016;18(8):839-850.
https://doi.org/10.1038/ncb3386
PMid:27398910 PMCid:PMC5040511 |
|
| |
20 Schröder M, Kaufman RJ. The
mammalian unfolded protein response. Annu Rev Biochem 2005;74:739-789.
https://doi.org/10.1146/annurev.biochem.73.011303.074134
PMid:15952902 |
|
| |
21 Rendu J, Montjean R, Coutton
C, Suri M, Chicanne G, Petiot A, Brocard J, Grunwald D, Pietri Rouxel F,
Payrastre B, Lunardi J, Dorseuil O, Marty I, Fauré J. Functional
characterization and rescue of a deep intronic mutation in OCRL gene
responsible for lowe syndrome. Hum Mutat 2017;38(2):152-159.
https://doi.org/10.1002/humu.23139
PMid:27790796 |
|
| |
22 Nakanishi K, Nozu K,
Hiramoto R, et al. A comparison of splicing assays to detect an intronic
variant of the OCRL gene in Lowe syndrome. Eur J Med Genet
2017;60(12):631-634.
https://doi.org/10.1016/j.ejmg.2017.08.001
PMid:28803024 |
|
| |
23 Abdalla E, El-Beheiry A,
Dieterich K, Thevenon J, Fauré J, Rendu J. Lowe syndrome: a particularly
severe phenotype without clinical kidney involvement. Am J Med Genet A
2018;176(2):460-464.
https://doi.org/10.1002/ajmg.a.38572
PMid:29226564 |
|
| |
24 Murakami Y, Wataya-Kaneda M,
Iwatani Y, Kubota T, Nakano H, Katayama I. Novel mutation of OCRL1 in Lowe
syndrome with multiple epidermal cysts. J Dermatol 2018;45(3):372-373.
https://doi.org/10.1111/1346-8138.13881
PMid:28516463 |
|
| |