
Citation: Kobat SG, Gul FC, Yusufoglu E. Bietti crystalline
dystrophy and choroidal neovascularization in childhood. Int J Ophthalmol 2019;12(9):1514-1516
DOI:10.18240/ijo.2019.09.24
・Letter to the Editor・
Bietti
crystalline dystrophy and choroidal neovascularization in childhood
Sabiha Gungor Kobat, Fatih Cem Gul,
Elif Yusufoglu
Department
of Ophthalmology, Elazig Health Sciences University, Elazig 23000, Turkey
Correspondence
to: Sabiha Gungor
Kobat. Department
of Ophthalmology, Elazig Health Sciences University, Elazig 23000, Turkey.
drsabihag@gmail.com
Received:
DOI:10.18240/ijo.2019.09.24
Citation:
Kobat SG, Gul FC, Yusufoglu E. Bietti crystalline dystrophy and choroidal
neovascularization in childhood. Int
J Ophthalmol
2019;12(9):1514-1516
Dear Editor,
I am Dr.
Sabiha Gungor Kobat, from the Department of Ophthalmology, Elazig Health
Sciences University, Elazig, Turkey. I am writing to present an exceedingly
rare case of two siblings, one of whom developed Bietti crystalline dystrophy
(BCD) with choroidal neovascularization at the age of 13 years, and the other
has asymptomatic BCD at 8 years old.
BCD, which
was first described by Bietti[1] in 1937, is
characterized by yellow-white deposits localized in the paralimbal cornea, and
superficial and deeper retinal layers[2-3]. Inheritance
is usually expressed as autosomal recessive but it can be found in autosomal
dominant cases[4]. As this slowly progressing
disease advances, atrophy, choroid sclerosis, and pigmental changes occur in
the retinal pigment epithelium (RPE). These result in a decrease in visual
acuity, night blindness, narrowing of the visual field, and central scotomas[2-3]. The development of choroidal neovascularization in
BCD is quite rare[5-12]. The
disease usually appears in the third decade of life, although cases have been
reported in the first or second decades[2,13].
The aim of this paper is to present an exceedingly rare case of two siblings,
one of whom developed BCD with choridal neovascularization at 13 years old,
while the other has asymptomatic BCD at 8 years old.
The report is in accordance with the Declaration of
Helsinki Ethical Principles. The patient has given her consent for her images
and related clinical information to be reported in this journal.
Case
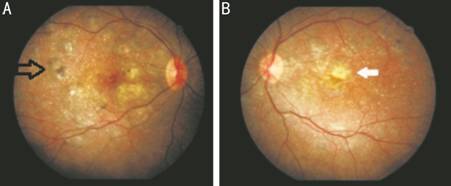
Figure 1
Color fundus pictures depicting the intraretinal crystals, foveal scar (white
arrow) and pigment clusters (black arrow)
A: Right eye; B: Left eye.

Figure 2
Common areas of chorioretinal atrophy in fluorescein angiography A: Right eye; B: Left eye.

Figure 3 OCT
of the right-left eye showing high reflective crystalline deposits
in the sensorineural retina and RPE, irregularity and increased reflectivity in
RPE A: Outer segment tabulation; B:
Subretinal scarring possibly due to old choroidal neovascularization.
Case 2 An 8-year-old male patient was
examined, and visual acuity was evaluated as

Figure 4 Colour fundus images A: Right eye; B: Left eye.
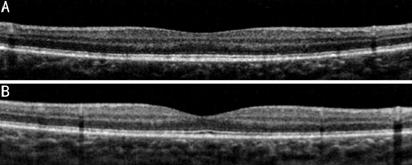
Figure 5 OCT images A: Right eye; B: Left eye.
BCD is a
rare, chorioretinal degeneration that is usually inherited through autosomal
recessive CYP4V2 (cytochrome P450 family 4 subfamily V polypeptide 2) gene due
to gene mutation. Usually dystrophy appears in the third decade of life, but it may also
occasionally be seen in the first or second decades[2,13]. BCD can result in legal blindness with decreasing
visual acuity and narrowing of the visual field in the fifth or sixth decades.
Choroidal neovascularization, cystoid macular edema, and macular hole have been
reported in patients with BCD[5-12,14-16]. Fuerst et al[12] treated a 34-year-old patient diagnosed with
choroidal neovascularization using bevacizumab. Nachiappan et al[9] diagnosed BCD which was complicated with choroidal
neovascular membrane (CNVM) in a 33-year-old male patient. The CNVM was treated
with intravitreal ranibizumab, and it was thought that the CNVM development may
have been due to crystalline irritation against the Bruch membrane[9]. Mamatha et al[8]
also diagnosed BCD which was complicated with CNVM in a 31-year-old patient,
and treated the CNVM with intravitreal ranibizumab. Atmaca et al[5] reported the case of a 13-year-old patient with CNVM in
the peripapillary region and serous retinal detachment in the fovea. Gupta et
al[6] diagnosed CNVM complicated by
juxtafoveal CNVM in a 64-year-old male patient. The lesion was inactive so was
not treated. The current patient was a 13-year-old girl. Numerous white-yellow
deposits, chorioretinal atrophic areas, and scar formation were detected, which
could have developed after CNVM. There were no findings of active CNVM such as
hemorrhagia or subretinal fluid. The OCT images showed high reflective
crystalline deposits in the sensorineural retina and RPE, and irregularity and
increased reflectivity in RPE. Subretinal scarring possibly due to old choroidal
neovascularization was detected in the left eye. There was no fluid in the
lesion. Although early studies showed crystals in the cornea to be diagnostic
criteria for BCD, later studies did not show corneal crystals in some patients.
In the current patient, no corneal crystals were observed. The presence of
common chorioretinal atrophy and scarring suggested that the disease had been
present for a long time. The patient’s mother and two siblings were also
examined carefully. No signs of the disease in the 19-year-old sister of the
patient whereas in the 8-year-old brother, crystals were detected in the retina
but not in the cornea. To the best of our knowledge, there are no cases in
literature of patients younger than this with BCD.
With careful
ophthalmological examination, BCD can be easily diagnosed. Diagnosis can be
supported by OCT and FFA findings. It was not possible to perform
electrophysiological and genetic testing on these cases because of the lack of
available facilities. The consanguinity of the parents of these patients
suggests that the genetic transition may be inherited through autosomal
recessive. This demonstrates that genetic transitions should be considered in
consanguineous marriages and cases can be diagnosed at a much younger age by conducting
health screening on all family members.
ACKNOWLEDGEMENTS
Conflicts of
Interest: Kobat
SG, None; Gul FC, None; Yusufoglu E, None.
REFERENCES