
・Basic
Research・
Effects
of epidermal growth factor on transforming growth factor-beta1-induced
epithelial-mesenchymal transition and potential mechanism in human corneal
epithelial cells
Shu-Yang
Chen1,2, Chen Xie1,3, Hong Zhu1,3, Ye Shen1
1Department of Ophthalmology, First
Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou 310003,
Zhejiang Province, China
2Tongde Hospital of Zhejiang
Province, Hangzhou 310012, Zhejiang Province, China
3Clinical Research Center, the First
Affiliate Hospital, School of Medicine, Zhejiang University, Hangzhou 310003,
Zhejiang Province, China
Co-first authors: Shu-Yang Chen and Chen Xie
Correspondence
to: Ye Shen.
Department of Ophthalmology, First Affiliated Hospital, College of Medicine,
Zhejiang University, 79 Qingchun Road, Hangzhou 310003, Zhejiang Province,
China. idrshen@zju.edu.cn
Received: 2019-07-10
Accepted: 2019-11-17
Abstract
AIM: To evaluate the
effects of epidermal growth factor (EGF) on transforming growth factor-beta1
(TGF-β1)-induced
epithelial-mesenchymal transition (EMT) in human corneal epithelial cells
(HCECs).
METHODS: HCECs were cultured
and treated with TGF-β1 for establishing the model of EMT in
vitro. Biological effect of EGF on TGF-β1-induced EMT was
evaluated. Proteins and mRNAs expression changes of E-cadherin, N-cadherin and
Fibronectin (EMT-relative markers) after TGF-β1 or TGF-β1 combined EGF
treatment were detected by Western blot and RT-PCR, respectively. Viability and
migration of HCECs were measured by CCK-8, transwell cell migration assay and
cell scratch wound healing assay. Activation of Smad2, ERK, p38, JNK and Akt
signaling pathways were evaluated by Western blot. Inhibitors of relevant
signaling pathways were added to the HCECs to explore the key signal mechanism.
RESULTS: With treatment of TGF-β1 only, three
EMT-relative proteins and mRNA expression showed that EMT up-regulated in a
concentration-dependent and time-dependent manner, with significantly
decreasing cell viability (TGF-β1≥5 ng/mL, P<0.05) and increasing
cell migration (TGF-β1≥5 ng/mL, P<0.01). The
phosphorylation of Smad2 and p38 was a key process of TGF-β1-induced EMT.
Meanwhile, EMT-relative proteins and mRNA expression showed that EGF inhibited
TGF-β1-indued EMT, with
significantly increasing cell viability (EGF≥10 ng/mL, P<0.01). It
was noteworthy that EGF significantly enhanced cell migration although EMT was
inhibited (EGF≥10 ng/mL, P<0.01), and the blockage of p38 (by
SB202190, a p38 inhibitor) was a potential mechanism of this phenomenon.
CONCLUSION: EGF inhibits TGF-β1-induced EMT via
suppressive p38, and promotes cells proliferation and migration in a non-EMT
process by inhibiting p38 pathway.
KEYWORDS: epidermal growth
factor; p38; epithelial-mesenchymal transition; corneal epithelial cell
DOI:10.18240/ijo.2020.01.03
Citation:
Chen SY, Xie C, Zhu H, Shen Y. Effects of epidermal growth factor on
transforming growth factor-beta1-induced epithelial-mesenchymal transition and
potential mechanism in human corneal epithelial cells. Int J Ophthalmol 2020;13(1):11-20
INTRODUCTION
The first layer of human corneal
structure is primarily composed of human corneal epithelial cells that play a
vital role in maintaining the integrity of ocular surface[1-2]. Both traumatic and pathologic factors will destroy the
structural integrity, thus leading to ophthalmodynia, tearing, foreign body
sensation and decreased visual quality[3-4].
A well-timed corneal epithelium reconstructionis required for symptom relieving
and better prognosis. Numerous researches demonstrate that
epithelial-mesenchymal transition (EMT) is a potential mechanism in ophthalmic
diseases progression as well as damage repair, including corneal epithelium
healing[5-6].
EMT, a progress of phenotypic
changes from epithelial cells to mesenchyme type, is known as a mechanism
related to embryonic development, wound healing, tissue fibrosis and cancer
progression[7]. The transition progress is induced
by several key transcription factors such as Snail, zinc finger E-box-binding
homeobox1 (ZEB1), ZEB2 and some other basic helix-loop-helix factors[7-10]. Certain signaling pathways have
been testified to get involved such as transforming growth factor-β (TGF-β),
epidermal growth factor (EGF), bone morphogenetic protein (BMP), fibroblast growth
factor (FGF), Wnt/β-catenin, insulin-like growth factor (IGF), etc.[11]. Among these, the TGF-β family are the most
significant and well-demonstrated. TGF-β induces EMT via both Smad and
non-Smad signaling pathways, the latter include ERK, p38, JUN N-terminal kinase
(JNK), Akt, etc.[11-12].
After being wounded, the cornea
shows a higher expression of TGF-β than normal situation, and up-regulates
epithelial cells migration and proliferation as a result. Clinically, the
impairment of cornea is better to be cured at the first stage. However, not all
of ophthalmic drug will bring advantages for corneal wound healing, because the
interaction between TGF-β and other factors remain unclear. According to
published studies, EGF has opposite effects in different tissues. One side, for
example, EGF can promote TGF-β-induced EMT in lung and pancreatic cancer cells[13]. On the other hand, EGF can suppress and even reverse
TGF-β-induced transition in normal hepatic cells[14].
In ocular tissues, sometime EGF and its analogues will be used in clinical
therapy. Such as recombinant EGF, which is beneficial to ocular surface defects
healing[15-17]. Nevertheless,
the effect and mechanism of EGF combining TGF-β remain unclear. The aim of our
research is to figure out the exact effect of EGF on TGF-β-induced EMT in human
corneal epithelial cells, and the signaling pathways potentially involved.
MATERIALS AND METHODS
Culture of Human Corneal Epithelial
Cells In our research, the human corneal
epithelial cell (HCEC) line from the Riken cell bank was used. The basal
culture medium was Dulbecco’s modified Eagle’s/Ham’s 12 medium (DMEM/F12),
containing heat-inactivated (56℃, 0.5h)
10% fetal bovine serum (FBS), 15 mmol/L HEPES buffer, penicillin (100 U/mL) and
streptomycin (100 mg/mL) (Gibco Company). The HCECs were cultured in constant
temperature incubator at 5%CO2, 95% air, 37℃
atmosphere, and passaged by trypsin/EDTA every 2-3d (the doubling time of the
HCECs with this condition was around 24h). During the experiment, the HCECs were
seeded in 6-well plates and Transwell chambers.
Study Design and Study Groups The study design is mainly divided
into three parts: 1) Establishing the TGF-β1 induced EMT models with the
concentration of 10 ng/mL for various periods of time (1, 3, 6d) or with
different concentrations (0, 1, 2, 5, 10, 20 ng/mL) for 2d. Evaluation of
EMT-relative markers protein and mRNA expression, cell viability and mobility.
2) Analysing Signaling pathways (Smad2, ERK, p38, JNK and Akt) and exploring
the possible signaling pathways involved in TGF-β1 induced EMT of HCECs. 3)
Exploring the biological functions of EGF (5, 10, 20 ng/mL) on the TGF-β1 (10
ng/mL) induced EMT, including three EMT-relative markers protein and mRNA
expression, cell viability and mobility. Furtherly, exploring the potential
mechanism and signaling pathway involved. The study groups were defined as
followed: 1) control group, treated with normal culture medium; 2) TGF-β1
treated groups; 3) TGF-β1 (10 ng/mL) combined with signal inhibitors (SB431542,
Smad2 inhibitor, 10 μmol/L; PD98059, ERK inhibitor, 20 μmol/L; SB202190, p38
inhibitor, 10 μmol/L; SP600125, JNK inhibitor, 10 μmol/L; Wortmannin, Akt
inhibitor, 1 μmol/L) treated groups; 4) EGF (5, 10, 20 ng/mL) treated groups;
5) EGF (5, 10, 20 ng/mL) combined with TGF-β1 (10 ng/mL) treated groups.
Cell Viability Assay Cell counting kit-8 (CCK-8; Dojindo
Laboratories, Kumamoto, Japan) had been used to evaluate the viability of HCECs
in different groups. Living cells can turn colorless WST-8 into soluble WST-8
formazan. The latter can be measured at the wavelength of 450 nm and the
density is in proportion to the content of metabolically normal cells.
RNA Extraction and Real-time
Quantitative PCR Total RNA was isolated from HCECs by
TRIzol reagent (Takara, Dalian, China), and then reverse transcribed to cDNA by
an RT reagent kit (Takara, Dalian, China). The sequences of the PCR primers
were Fibronectin (forward: 5-CGGTGGCTGTCAGTCAAAG-3; reverse:
5-AAACCTCGGCTTCCTCCATAA-3), N-cadhein (forward: 5’-TCAGGCGTCTGTAGAGGCTT-3’;
reverse: 5’-ATGCACATCCTTCGATAAGACTG-3’), E-cadherin (forward: 5’-AAAGGCCCATTTCCTAAAAACCT-3’; reverse: 5’-TGCGTTCTCTATCCAGAGGCT-3’),
GAPDH (forward: 5’-GGCCTCCAAGGAGTAAGACC-3’; reverse: 5’-AGGGGTCTACATGGAAACTG-3’).
The primers above were provided from Sango Biotech (Shanghai, China). The cDNA
combining primers and other required ingredients was amplified and evaluated by
an ABI 7500 real-time PCR system (Applied Biosystems, Carlsbad, USA).
Western Blot Analysis The total cellular protein was
extracted by cell total protein extraction kit (Sangon, Shanghai, China). Same
amount of protein lysates (volumes at 20-30 μL) were electrophoresed in 10% or
20% sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE; Bio-Rad), and then
transferred (at 4℃, 350 mA for 2h) to
polyvinylidene difluoride (PVDF) membranes. After blocked with 5% nonfat milk
or 5% bovine serum albumin (BSA) in Tris-buffered saline with 0.1% Tween-20
(TBST), the transferred PVDF membranes were incubated with primary antibodies
at 4℃
overnight, and incubated with second antibody at room temperature for 60min.
The results were detected by chemiluminescence reagents (ECL, Millipore) and
exposed by an imaging system (ChemiDoc MP, Bio-Rad).
Transwell Cell Migration Assay Cell migration was performed in
Transwell chambers with polycarbonate membrane, 8.0 μm pore size. HCECs (5×104
cells/mL) were combined with 200 μL FBS-free medium with or without TGF-β1 or
EGF, and then added to the upper chamber. Medium 600 μL with 10% FBS were added
to the lower chamber. The system was incubated for 48h at 37℃.
The cells on the upside of the filters were removed by cotton swab. The cells
on the underside of the filters were fixed by 75% methyl alcohol and stained by
crystal violet. Cells were counted to evaluate the migration across the
filters.
Cell Scratch Wound Healing
Assay HCECs (1.5×105 cells/mL)
were seeded into 6-well plates, with DMEM/F12 containing 10% FBS. After 48h,
the monolayer was scratched with 1 mL pipette tip. The instant and 24-hour photomicrographs
were taken (Nikon ECLIPSE Ti, Japan), and measured by an imaging analyzing
software (NIS Elements 4.0, Nikon, Japan).
Statistical Analysis Each result was experimented
independently at least three times and was given as mean±standard deviation
(SD). Data were analyzed by one-way ANOVA test with Bonferroni correction in
comparing data from more than two groups and Student’s t-test in
comparing data between two groups. Data analyzing was carried out by SPSS
Statistics (Version 21.0, SPSS Inc., USA). P<0.05 indicated
statistical significance.
RESULTS
Effects of TGF-β1 on HCECs
Epithelial-mesenchymal Transition, Proliferation and Migration E-cadherin, N-cadherin, and Fibronectin[18] were detected by Western blot (Figure 1). HCECs were
treated with TGF-β1 of 1, 2, 5, 10, 20 ng/mL for 2d. As shown in Figure 1A, E-cadherin significantly down-regulated
in 10 and 20 ng/mL (P<0.05); N-cadherin and Fibronectin significantly
up-regulated in all concentration and peaked around 5-10 ng/mL (N-cadherin, P<0.01;
Fibronectin, P<0.001). According to the results, we chose TGF-β1 at
10 ng/mL for the subsequent experiments. The time course of 1, 3, 6d with
TGF-β1 at 10 ng/mL was also treated on HCECs. As shown in Figure 1B, N-cadherin,
Fibronectin significantly increased at 1d (N-cadherin, P<0.01;
Fibronectin, P<0.001), and E-cadherin significantly decreased at 3
and 6d (P<0.05, P<0.01).
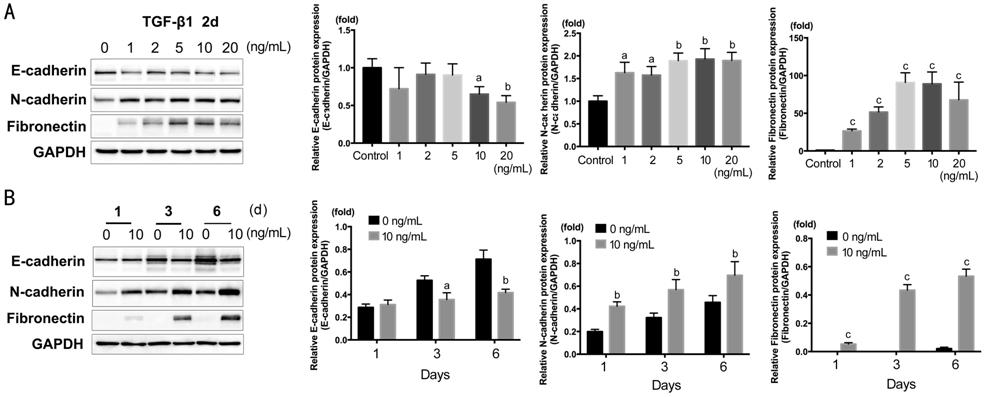
Figure 1 Western blot showing the
dose response and time course of E-cadherin, N-cadherin and Fibronectin actived
by TGF-β1 in HCECs A: TGF-β1 concentration gradient of 0,
1, 2, 5, 10, 20 ng/mL in 2d; B: Time course of 1, 3, 6d with TGF-β1 at 10
ng/mL. aP<0.05, bP<0.01, cP<0.001
vs control group.
We also demonstrated the mRNA
expression in Figure 2. The result of N-cadherin and Fibronectin was the same
to protein expression, showing increasing concentration-dependence and
time-dependence. But the E-cadherin mRNA showed stimulated rather than
decreasing as above. The remarkable stimulation of E-cadherin started from 10
ng/mL, and after 3d declined to the level of TGF-β1 free group. This indicate
that the regulation of E-cadherin was after transcriptional level in
TGF-β1-induced EMT.
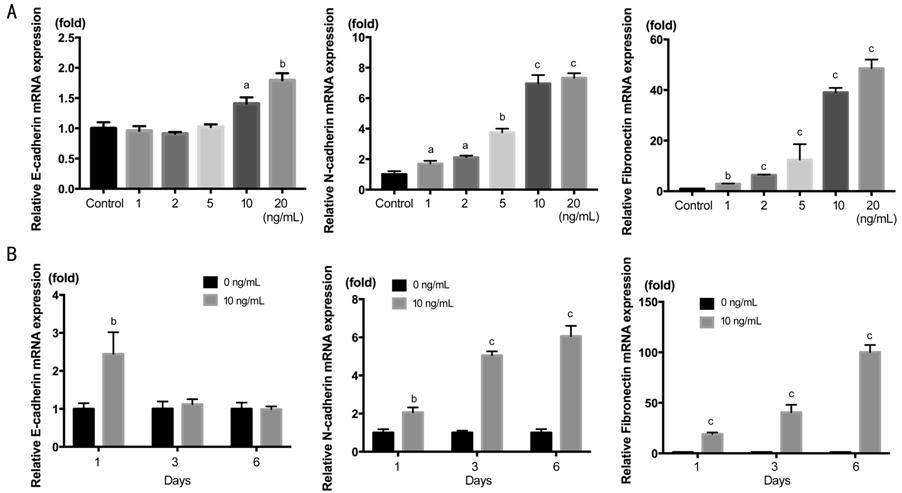
Figure 2 RT-PCR showing the dose
response and time course of E-cadherin mRNA, N-cadherin and Fibronectin mRNA
actived by TGF-β1 in
HCECs A: TGF-β1 concentration gradient of
0, 1, 2, 5, 10, 20 ng/mL in 2d; B: Time course of 1, 3, 6d with TGF-β1 at 10
ng/mL. aP<0.05, bP<0.01, cP<0.001
vs control group.
The cell viability assay was
detected by CCK-8 (Figure 3A),
and showed inversely concentration-dependent manner from 5 ng/mL with TGF-β1
treatment (P<0.05). TGF-β1 inhibited the proliferation of HCECs. The
transwell assay (Figure 3B) revealed that TGF-β1 promoted cell migration with
concentration-dependent manner. All concentration showed significant
increasing, and peaked at 10 ng/mL (P<0.001). Cell scratch wound
healing assay was the consistent result (Figure 3C) to transwell. After TGF-β1 (10 ng/mL) exposure for
24h, the greater migration distance of HCECs was remarkable (P<0.001).
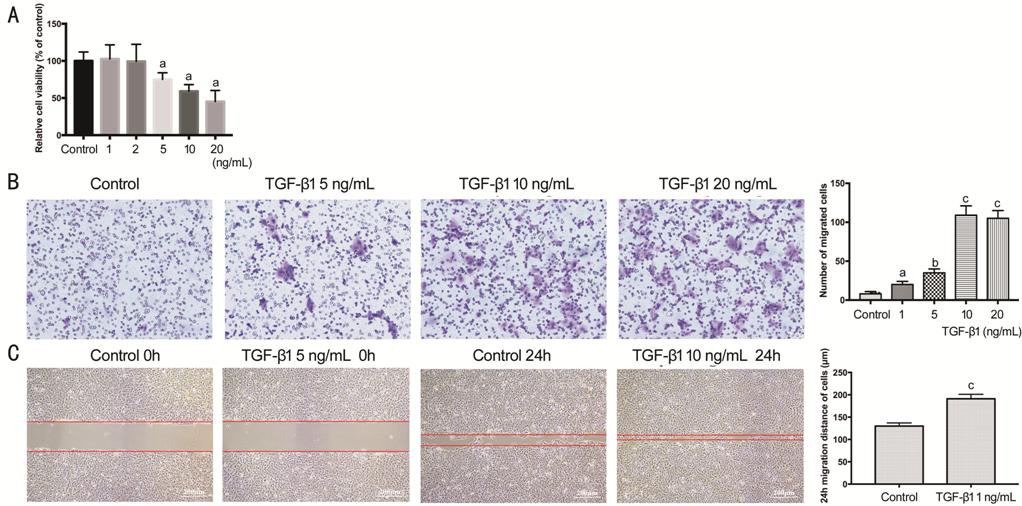
Figure 3 Viability and migration of
HCECs treated with TGF-β1 A: The viability of HCECs actived by TGF-β1
concentration gradient of 0, 1, 2, 5, 10, 20 ng/mL in 2d, expressed as a
percentage of the control group; B: HCECs migration in transwell assay; C:
HCECs migration in cell scratch wound healing assay. aP<0.05,
bP<0.01, cP<0.001 vs control
group.
Signaling Pathways Involved in
TGF-β1-induced Epithelial-Mesenchymal Transition The activation of Signaling pathways
were detected by Western blot (Figure 4A).
The results showed significant phosphorylated of Smad2 and p38. The maximal
expression presented at 30min for p-Smad2 (P<0.001), and at 60min for
p-p38 (P<0.001). In contrast, p-ERK1/2, p-JNK, p-Akt remained
unchanged. In the meantime, with the treatment of TGF-β1 (10 ng/mL) combining
Smad2 inhibitor (SB431542), ERK inhibitor (PD98059), p38 inhibitor (SB202190),
JNK inhibitor (SP600125), and Akt inhibitor (Wortmannin) for 2d. According to
the result of Fibronectin and N-cadherin, the inhibition of Smad2, ERK, p38 and
JNK would distinctly suppress TGF-β1-induced EMT; on the contrary, the
inhibition of Akt promote EMT (Figure 4B). The change of E-cadherin was less
than Fibronectin and N-cadherin, especially the mRNA expression which did not
show any significant difference.
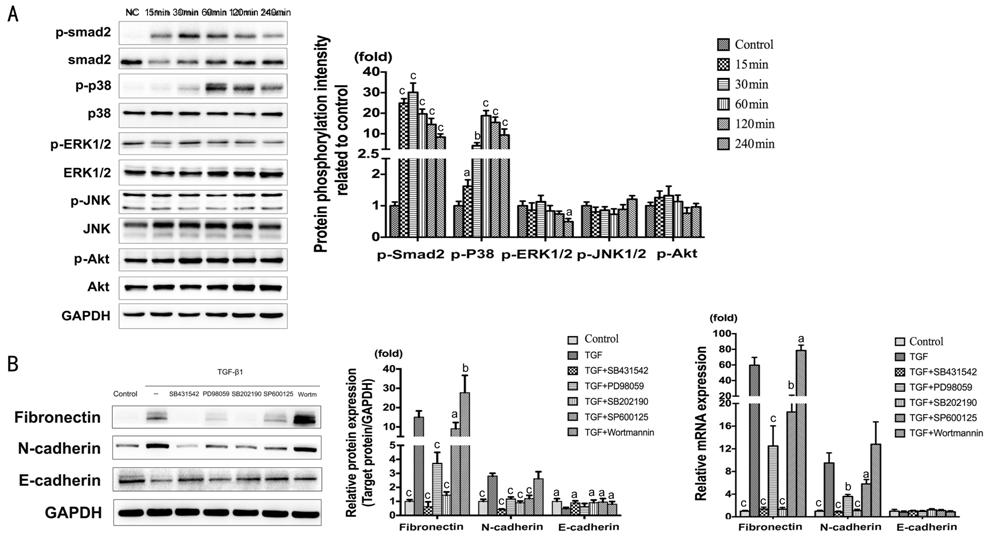
Figure 4 Signaling pathways involved
in TGF-β1-induced EMT A: The expression of
p-Smad2/Smad2, p-p38/p38, p-ERK/ERK, p-JNK/JNK and p-Akt/Akt at 0, 15, 30, 60,
120, 240min, treated with TGF-β1 (10 ng/mL) in HCECs. aP<0.05,
bP<0.01, cP<0.001 vs control
group. B: The expression of Fibronectin, N-cadherin and E-cadherin treated with
TGF-β1 (10 ng/mL) combining Smad2 inhibitor (SB431542, 10 μmol/L), ERK inhibitor
(PD98059, 20 μmol/L), p38 inhibitor (SB202190, 10 μmol/L), JNK inhibitor
(SP600125, 10 μmol/L), and Akt inhibitor (Wortmannin, 1 μmol/L) for 2d. aP<0.05,
bP<0.01, cP<0.001 vs TGF-β1
group.
The proliferation and migration of
HCECs were taken into consideration (Figure 5). The cell viability assay
(CCK-8) showed that the inhibition of ERK and JNK pathways significantly
suppress the proliferation of HCECs (P<0.01), but the inhibition of
Smad2 and p38 pathways was positive impact compared with TGF-β1 group (P<0.05;
Figure 5A-5B). As
for the cell migration assay (transwell), the inhibition of Smad2, ERK, JNK and
Akt decreased the migration of HCECs, and it deserved to be mentioned that the
inhibition of p38 prominently increased cell migration (P<0.01).
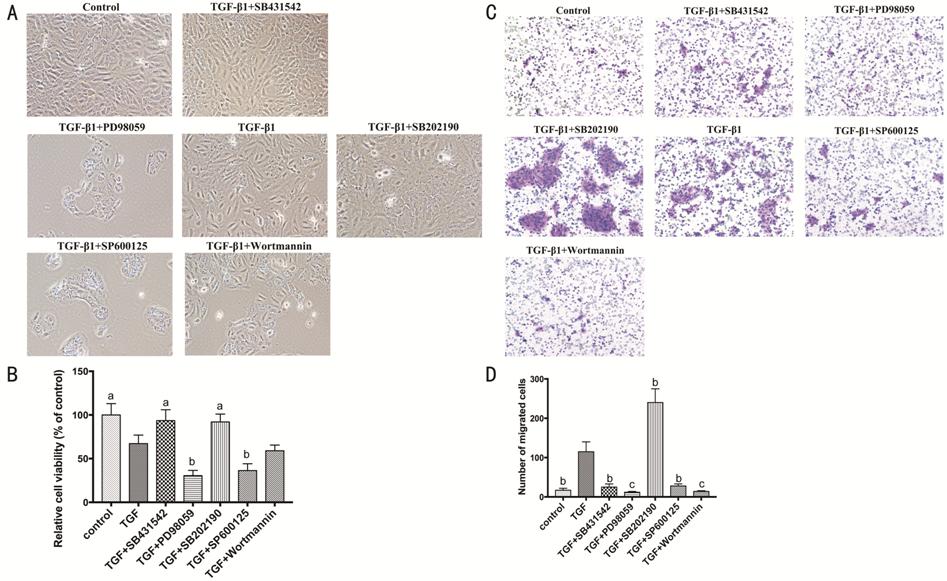
Figure 5 Signaling pathways involved
in proliferation and migration of HCECs A: HCECs treated with TGF-β1 (10 ng/mL)
combining Smad2 inhibitor (SB431542, 10 μmol/L), ERK inhibitor (PD98059, 20
μmol/L), p38 inhibitor (SB202190, 10 μmol/L), JNK inhibitor (SP600125, 10
μmol/L), and Akt inhibitor (Wortmannin, 1 μmol/L); B: The viability of HCECs;
C-D: HCECs migration in transwell assay. aP<0.05, bP<0.01,
cP<0.001 vs TGF-β1 group.
Effect of EGF on TGF-β1-induced
Epithelial-Mesenchymal Transition, Proliferation and Migration In comparison to TGF-β1 (10 ng/mL)
group, Fibronectin and N-cadherin showed evident low expression in the groups
with EGF (5, 10, 20 ng/mL, with or without TGF-β1; P<0.01), while
E-cadherin showed up-regulation of protein level (P<0.05) and
remained unchanged of mRNA expression. Concentration-dependence was not obvious
(Figure 6).
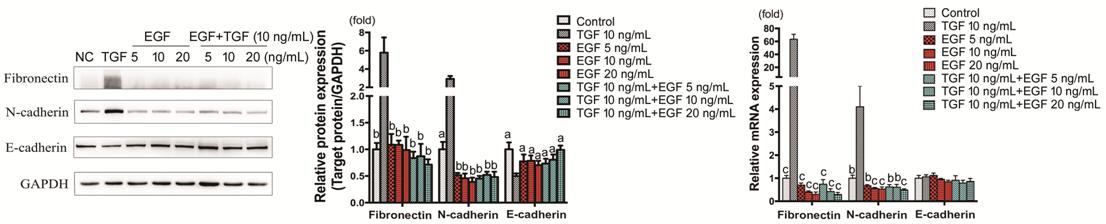
Figure 6 Fibronectin, N-cadherin and
E-cadherin expression in HCECs treated with TGF-β1 and EGF HCECs were treated with TGF-β1, EGF, or
TGF-β1+EGF and Fibronectin, N-cadherin and E-cadherin protein and mRNA
expressions were detected by Western blot and RT-PCR respectively. aP<0.05,
bP<0.01, cP<0.001 vs TGF-β1
group.
The proliferation of HCECs treated
with EGF (10 ng/mL) was promoted (P<0.001; Figure 7A). A cluster of cells like huge islands
could be seen in EGF group and EGF+TGF group (P<0.001). The cell
viability showed that EGF enhanced proliferation of HCECs with
concentration-dependence, and could reverse the suppressive effect of TGF-β1 on
cells proliferation (Figure 7B). Cells migration showed that EGF (10 ng/mL)
remarkably promoted HCECs’ migration. Interestingly, the combinative group of
TGF-β1 (10 ng/mL) and EGF (10 ng/mL) developed more migration capability than
the groups with TGF-β1 or EGF only (P<0.001; Figure 7C, 7D).
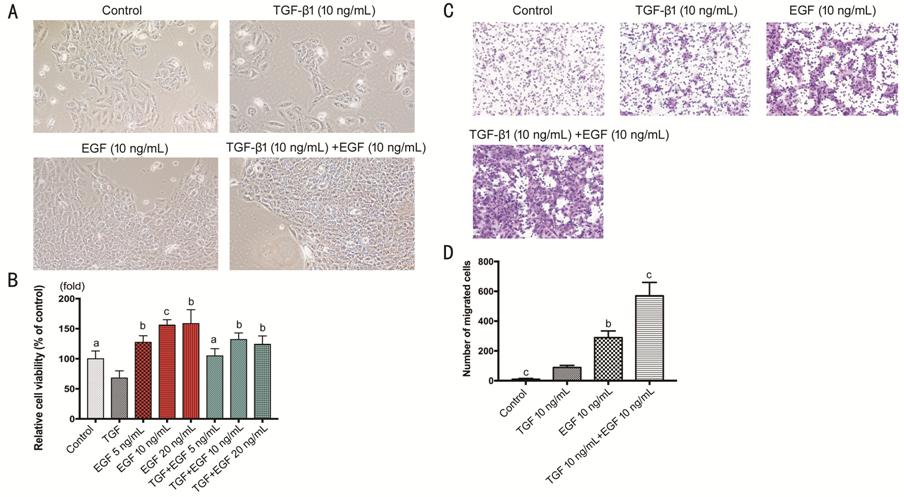
Figure 7 Effect of EGF on
TGF-β1-induced proliferation and migration of HCECs A: HCECs treated with TGF-β1, EGF, or
TGF-β1+EGF; B: The viability of TGF-β1 treated HCECs with or without EGF; C-D:
HCECs migration in transwell treated with TGF-β1 with or without EGF. aP<0.05,
bP<0.01, cP<0.001 vs TGF-β1
group.
Effect of EGF on Signaling Pathways
in TGF-β1-induced Epithelial-Mesenchymal Transition The phosphorylation of Akt, ERK, p38 and
Smad2 in HCECs was detected after
TGF-β1 and EGF treatment (Figure 8). For p38 Signaling pathway, TGF-β1 brought
a significant promotion, but EGF remarkably blocked this effect. The blockage
of EGF was more obvious at 2h, approaching the control group. The activation of
Smad2 signaling pathway induced by TGF-β1 was quite strong (over 30 times of
control group), and was also inhibited by EGF, but the inhibition could not be
detected until 2h. ERK signaling pathway was activated in groups with EGF, and
the group with both TGF-β1 and EGF showed stronger activation, especially at
1h. As for Akt signaling pathway was inhibited in groups with EGF, and the
inhibition was more significant in 2h group.
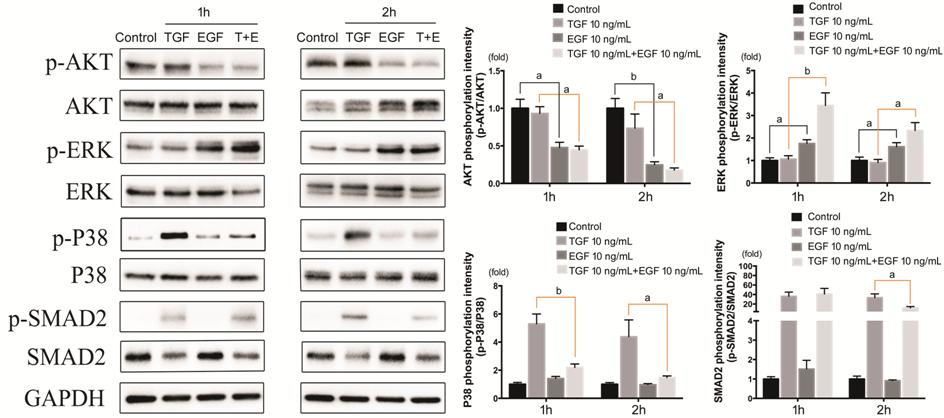
Figure 8 Effect of EGF on signaling
pathways in TGF-β1-induced EMT The
expression of p-Akt/Akt, p-ERK/ERK, p-p38/p38, p-Smad2/Smad2 in HCECs treated with TGF-β1 and EGF. aP<0.05,
bP<0.01, cP<0.001.
DISCUSSION
There are three isoforms of TGF-β
(TGF-β1, TGF-β2 and TGF-β3). TGF-β1 is at a low level in normal cornea, and
after wounding it will show a high level along the Bowman’s layer. TGF-β2 keeps
prominent expression in normal cornea, and up-regulates in epithelial cells
migration after wounding. TGF-β3 is observed only in the basal cells, and
showed weak relationship to epithelial cells migration[19].
In this study, we chose TGF-β1 in
order to figure out the mechanism of TGF-β-induced EMT.
In the present study, we observed
that TGF-β1 presented promotion of EMT in HCECs, on both levels of mRNA and
protein (Figures 1, 2). And it showed suppressive proliferation and enhanced
migration of cells (Figure 3). In lots of studies, TGF-β family proteins are
well demonstrated to be the inducer of EMT[20],
not only in ocular tissues, but also in many other organs. TGF-β is implicated
in numerous pathological processes such as carcinogenesis, immoderate tissue
fibrosis, and immunological disorders. TGF-β plays a profitable role in some
physiological activities[21]. For example,
investigators reveal that in early stages of cancer, TGF-β shows tumor
suppressive effects, but in the late stages, TGF-β promotes invasiveness and
metastasis[22]. For eyes, TGF-β exerts influence
on both sides too, participating in inflammation, fibrosis, or acting as a
helper for wound healing[23].
Signaling pathways are key processes
for TGF-β exerting effect. In the control group and at the very beginning of
the treatment by TGF-β1, the expression of p-Smad2 and p38 was at an
extraordinary low level. As time course going on, these two pathways showed
peak level at 30 and 60min (Figure 4A).
It indicated that TGF-β1 acted as the trigger of p-Smad2 and p38 pathways’
activation. In numbers of researches, TGF-β is well demonstrated to be an
important participator of EMT via Smad or non-Smad pathways[24-25]. Smad2/3 are key signaling
molecules that are phosphorylated after TGF binding to TGF receptor. In this
process, lots of Smads participate in Smad-depending signaling, such as
coactivator Smad4, inhibitory regulator Smad6 and Smad7[11,26]. The non-Smad pathways consist of many
Smad-independent signaling, like p38, ERK, JNK and Akt, as we selected in this
study. Some researchers mention that there are certain interactions between
Smad and non-Smad pathways. For instance, p38 pathway activates phosphorylation
of Smad3 thus leading to the enhancement of Smad3/4 complex formation[27].
The treatment of inhibitors revealed
the parallel conclusion. When Smad2 and p38 pathways were blocked, EMT was
inhibited on mRNA and protein levels (Figure 4B), and cells proliferation
increased (Figure 5A, 5B). As
for the cells migration (Figure 5C,
5D), Smad2’s
inhibition showed down-regulation as mentioned, but p38 was a little different.
The blockage of p38 brought a high promotion of migration in HCECs like EMT
process, however the EMT-relative mRNA and protein expression was decreased.
Researchers investigate that inhibition of p38 partially reverses EMT changes
in breast cancer cells, with decreasing gene expression of the EMT markers
Twist, Snail, Slug and ZEB, as well as N-cadherin protein[28].
And p38 MAPKs have been implicated in phosphorylation of serine 68 which is a
major phosphorylation site of Twist1, thus promoting EMT[29].
Moreover, our study revealed that the inhibition of p38 pathway would promote
cellular viability and migration of HCECs, and this phenomenon has rarely been
mentioned. In cardiomyocytes, some research shows that the blockage of p38
signaling pathway can rescue the reduced cell viability[30].
The suppression of ERK and JNK also showed decreasing EMT-relative mRNA and
protein expression. However, the proliferation of HCECs was remarkably
inhibited (Figure 5A, 5B), and
it was considered to be an indispensable reason of decreasing EMT. In our point
of view, ERK and JNK are essential for HCECs’ survival, which is the foundation
of EMT’s initiation and development[31-33].
When the corneal epithelium is
injuried, growth factors (EGF, PDGF, TGF, FGF, IGF-I, KGF and HGF) and
inflammatory factors (IL-1, IL-6, and IL-10, and TNF-α) are released, which are
important regulators that stimulate corneal epithelial cell growth,
proliferation, migration, differentiation, adhesion involved in wound healing.
They also mediate different cell functions including intracellular and
intercellular signaling molecules. TGF-β stimulates corneal epithelial cell
migration via integrin β1, which mediates p38 MAPK activation,
extracellular matrix expression and EMT leading to increased cell mobility. The
fundamental purpose of this study was to figure out the effect of EGF on TGF-β1-induced
EMT and its potential mechanism within HCECs. We observed that EGF itself would
not induce EMT, and it could restrain TGF-β1-induced EMT on both mRNA and
protein levels, within the concentration from 5 to 20 ng/mL (Figure 6). The
suppressive effect of EGF on TGF-β-induced EMT has been reported in some organs
and tissues. EGF will restrain the initiation of TGF-β1-induced EMT, and even
reverse the transition[14]. Besides, some
researchers prove that EGF can induce phosphorylation and expression of
TGF-β/Smad repressor, thus inhibiting TGF-β1-stimulated markers of EMT[34].
Furthermore, EGF brought prominent
raising of HCECs’ proliferation (Figure 7A, 7B). The micrographs showed different cells
morphological characteristic in comparison to control group and TGF-β1 group.
HCECs treated with EGF tended to be clusters of cells like huge island, rather
than scattered. The exist of EGF relieved the suppressive impact of TGF-β1 on
HCECs’ proliferation.
In addition, the cells migration
with EGF was enhanced (Figure 7C,
7D). We observed that the EGF group showed higher migration than TGF-β1 group,
and the combing group with both EGF and TGF-β1 showed the highest. EGF
significantly inhibited TGF-β1-induced EMT in HCECs, but the cell motility
remarkably enhanced. This is an interesting discovery, because numerous
researches point out that EMT is positively correlated to cells motility and
migration[6,35]. In some
studies, it has been demonstrated that EGF promotes motility and migration of
cells including HCECs in a non-EMT way, for instance, through ROS, MEK/ERK and
JNK pathways[36-38]. In our
research, targeted inhibition of p38 brought decreasing effect on EMT, but the
proliferation and migration of HCECs increased. This phenomenon was parallel to
the effect of EGF combining TGF-β1. Furthermore, we found that EGF
significantly inhibited the phosphorylation of p38 (Figure 8), thus producing
biological effects. We suspect that p38 is a key signaling pathway for EGF to
inhibit TGF-β1-induced EMT and promote HCECs’ proliferation and migration.
We hold the opinion that EGF
inhibits TGF-β1-induced EMT via suppressive p38 and Smad2, and promotes
cells proliferation and migration in a non-EMT process by inhibiting
p38-pathway. The combination of EGF and TGF-β1 enhances HCECs’ proliferation
and migration, bringing advantage for corneal epithelium wound healing. It
reveals that EMT is not the only process for corneal epithelium healing. EGF
up-regulates the motility and migration of HCECs via p38 inhibition, in
a non-EMT manner.
ACKNOWLEDGEMENTS
Authors’ contributions: Chen SY completed the main part of
the experimental research; Xie C analyzed the experimental data, and was a
co-author in writing the manuscript; Zhu H completed part of this study, and
provided experimental materials; Shen Y designed the methods of this study. All
authors read and approved the final manuscript
Foundations: Supported by the 63th
Postdoctoral Science Foundation of China (No.2018M632487); General Natural Science Projects,
Department of Education, Zhejiang Province, China (No.Y201636718).
Conflicts of Interest: Chen SY, None; Xie C, None;
Zhu H, None; Shen Y, None.
REFERENCES
|
1 Mobaraki M, Abbasi R, Omidian Vandchali
S, Ghaffari M, Moztarzadeh F, Mozafari M. Corneal repair and regeneration: current
concepts and future directions. Front Bioeng Biotechnol 2019;7:135.
https://doi.org/10.3389/fbioe.2019.00135
PMid:31245365 PMCid:PMC6579817
|
|
|
|
2 Goswami DG, Kant R, Ammar DA, Kumar D,
Enzenauer RW, Petrash JM, Tewari-Singh N, Agarwal R. Acute corneal injury in
rabbits following nitrogen mustard ocular exposure. Exp Mol Pathol
2019;110:104275.
https://doi.org/10.1016/j.yexmp.2019.104275
PMid:31233733
|
|
|
|
|
3 Tang L, Wang X, Wu J, Li SM, Zhang Z, Wu
S, Su T, Lin Z, Chen X, Liao X, Bai T, Qiu Y, Reinach PS, Li W, Chen Y, Liu
Z. Sleep deprivation induces dry eye through inhibition of PPARα expression
in corneal epithelium. Invest Ophthalmol Vis Sci 2018;59(13):5494-5508.
https://doi.org/10.1167/iovs.18-24504
PMid:30658033
|
|
|
|
|
4 Yin J, Jurkunas U. Limbal stem cell
transplantation and complications. Semin Ophthalmol 2018;33(1):134-141.
https://doi.org/10.1080/08820538.2017.1353834
PMid:29172876
|
|
|
|
|
5 Kowtharapu BS, Stahnke T, Wree A, Guthoff
RF, Stachs O. Corneal epithelial and neuronal interactions: role in wound
healing. Exp Eye Res 2014;125:53-61.
https://doi.org/10.1016/j.exer.2014.05.006
PMid:24880142
|
|
|
|
|
6 Liu T, Dong XG. The progress of
epithelial-mesenchymal transition in ophthalmology. Zhonghua Yan Ke Za Zhi
2008;44(3):285-288.
|
|
|
|
|
7 Lamouille S, Xu J, Derynck R. Molecular mechanisms
of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol
2014;15(3):178-196.
https://doi.org/10.1038/nrm3758
PMid:24556840 PMCid:PMC4240281
|
|
|
|
|
8 Lin YW, Dong CF, Zhou BP. Epigenetic
regulation of EMT: the Snail story. Curr Pharm Des 2014;20(11):1698-1705.
https://doi.org/10.2174/13816128113199990512
PMid:23888971
|
|
|
|
|
9 Smith BN, Bhowmick NA. Role of EMT in
metastasis and therapy resistance. J Clin Med 2016;5(2):E17.
https://doi.org/10.3390/jcm5020017
PMid:26828526 PMCid:PMC4773773
|
|
|
|
|
10 Zhang PJ, Sun YT, Ma L. ZEB1: at the
crossroads of epithelial-mesenchymal transition, metastasis and therapy
resistance. Cell Cycle 2015;14(4):481-487.
https://doi.org/10.1080/15384101.2015.1006048
PMid:25607528 PMCid:PMC4614883
|
|
|
|
|
11 Gonzalez DM, Medici D. Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal
2014;7(344):re8.
https://doi.org/10.1126/scisignal.2005189
PMid:25249658 PMCid:PMC4372086
|
|
|
|
|
12 Miyazono K. Transforming growth factor-β
signaling in epithelial-mesenchymal transition and progression of cancer.
Proc Jpn Acad Ser B Phys Sci 2009;85(8):314-323.
https://doi.org/10.2183/pjab.85.314
PMid:19838011 PMCid:PMC3621568
|
|
|
|
|
13 Buonato JM, Lan IS, Lazzara MJ. EGF augments
TGFβ-induced epithelial-mesenchymal transition by promoting SHP2 binding to
GAB1. J Cell Sci 2015;128(21):3898-3909.
https://doi.org/10.1242/jcs.169599
PMid:26359300
|
|
|
|
|
14 Wang P, Yang AT, Cong M, Liu TH, Zhang
D, Huang J, Tong XF, Zhu ST, Xu Y, Tang SZ, Wang BE, Ma H, Jia JD, You H. EGF
suppresses the initiation and drives the reversion of TGF-β1-induced
transition in hepatic oval cells showing the plasticity of progenitor cells.
J Cell Physiol 2015;230(10):2362-2370.
https://doi.org/10.1002/jcp.24962
PMid:25739869
|
|
|
|
|
15 Yan L, Wu W, Wang Z, Li C, Lu X, Duan H,
Zhou J, Wang X, Wan P, Song Y, Tang J, Han Y. Comparative study of the
effects of recombinant human epidermal growth factor and basic fibroblast
growth factor on corneal epithelial wound healing and neovascularization in
vivo and in vitro. Ophthalmic Res 2013;49(3):150-160.
https://doi.org/10.1159/000343775
PMid:23258255
|
|
|
|
|
16 Wu W, Zeng LN, Peng YY, Lu XH, Li CY,
Wang ZC. The effects of recombinant human epithelialgrowth factor and protein-free
calf blood extract for recovery of corneal mechanical epithelial defects
healing and neovascularization. Eur Rev Med Pharmacol Sci
2014;18(22):3406-3411.
|
|
|
|
|
17 Li Z, Lin YS, Guo H, Li DM, Du YM, Zhang
HY. Effect of recombinant epidermal growth factor on ocular surface
re-epithelization following amniotic membrane transplantation in patients
with pterygium excision. Acad J First Med Coll PLA 2002;22(5):437-438.
|
|
|
|
|
18 Zeisberg M, Neilson EG. Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest 2009;119(6):1429-1437.
https://doi.org/10.1172/JCI36183
PMid:19487819 PMCid:PMC2689132
|
|
|
|
|
19 Huh MI, Chang Y, Jung JC. Temporal and
spatial distribution of TGF-beta isoforms and signaling intermediates in
corneal regenerative wound repair. Histol Histopathol 2009;24(11):1405-1416.
|
|
|
|
|
20 Ahmadi A, Najafi M, Farhood B, Mortezaee
K. Transforming growth factor-β signaling: tumorigenesis and targeting for
cancer therapy. J Cell Physiol 2019;234(8):12173-12187.
https://doi.org/10.1002/jcp.27955
PMid:30537043
|
|
|
|
|
21 Margadant C, Sonnenberg A.
Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep
2010;11(2):97-105.
https://doi.org/10.1038/embor.2009.276
PMid:20075988 PMCid:PMC2828749
|
|
|
|
|
22 Syed V. TGF-β signaling in cancer. J
Cell Biochem 2016;117(6): 1279-1287.
https://doi.org/10.1002/jcb.25496
PMid:26774024
|
|
|
|
|
23 Zubair M, Ahmad J. Role of growth factors
and cytokines in diabetic foot ulcer healing: A detailed review. Rev Endocr
Metab Disord 2019;20(2):207-217.
https://doi.org/10.1007/s11154-019-09492-1
PMid:30937614
|
|
|
|
|
24 Zi Z. Molecular engineering of the TGF-β
signaling pathway. J Mol Biol 2019;431(15):2644-2654.
https://doi.org/10.1016/j.jmb.2019.05.022
PMid:31121181
|
|
|
|
|
25 Derynck R, Budi EH. Specificity,
versatility, and control of TGF-β family signaling. Sci Signal
2019;12(570):eaav5183.
https://doi.org/10.1126/scisignal.aav5183
PMid:30808818 PMCid:PMC6800142
|
|
|
|
|
26 Saika S. TGFbeta pathobiology in the
eye. Lab Invest 2006;86(2):106-115.
https://doi.org/10.1038/labinvest.3700375
PMid:16341020
|
|
|
|
|
27 Furukawa F, Matsuzaki K, Mori S, Tahashi
Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki Y,
Nishizawa M, Fujisawa J, Inoue K. P38 MAPK mediates fibrogenic signal through
Smad3 phosphorylation in rat myofibroblasts. Hepatology 2003;38(4):879-889.
https://doi.org/10.1002/hep.1840380414
|
|
|
|
|
28 Antoon JW, Nitzchke AM, Martin EC,
Rhodes LV, Nam S, Wadsworth S, Salvo VA, Elliott S, Collins-Burow B, Nephew KP,
Burow ME. Inhibition of p38 mitogen-activated protein kinase alters microRNA
expression and reverses epithelial-to-mesenchymal transition. Int J Oncol
2013;42(4):1139-1150.
https://doi.org/10.3892/ijo.2013.1814
PMid:23403951 PMCid:PMC3622654
|
|
|
|
|
29 Hong J, Zhou J, Fu JJ, He T, Qin J, Wang
L, Liao L, Xu JM. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes
Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res
2011;71(11):3980-3990.
https://doi.org/10.1158/0008-5472.CAN-10-2914
PMid:21502402 PMCid:PMC3107354
|
|
|
|
|
30 Hu J, Xu X, Zuo Y, Gao X, Wang Y, Xiong C,
Zhou H, Zhu S. NPY impairs cell viability and mitochondrial membrane
potential through Ca2+ and p38 signaling pathways in neonatal rat
cardiomyocytes. J Cardiovasc Pharmacol 2017;70(1):52-59.
https://doi.org/10.1097/FJC.0000000000000493
PMid:28437279
|
|
|
|
|
31 Hu Y, Mintz A, Shah SR,
Quinones-Hinojosa A, Hsu W. The FGFR/MEK/ERK/brachyury pathway is critical
for chordoma cell growth and survival. Carcinogenesis 2014;35(7):1491-1499.
https://doi.org/10.1093/carcin/bgu014
PMid:24445144 PMCid:PMC4593008
|
|
|
|
|
32 Alexaki VI, Javelaud D, Mauviel A. JNK supports
survival in melanoma cells by controlling cell cycle arrest and apoptosis.
Pigment Cell Melanoma Res 2008;21(4):429-438.
https://doi.org/10.1111/j.1755-148X.2008.00466.x
PMid:18541008
|
|
|
|
|
33 Hossain MS, Ifuku M, Take S, Kawamura J,
Miake K, Katafuchi T. Plasmalogens rescue neuronal cell death through an
activation of AKT and ERK survival signaling. PLoS One 2013;8(12):e83508.
https://doi.org/10.1371/journal.pone.0083508
PMid:24376709 PMCid:PMC3869814
|
|
|
|
|
34 Liu X, Hubchak SC, Browne JA, Schnaper
HW. Epidermal growth factor inhibits transforming growth factor-β-induced
fibrogenic differentiation marker expression through ERK activation. Cell
Signal 2014;26(10):2276-2283.
https://doi.org/10.1016/j.cellsig.2014.05.018
PMid:24905473 PMCid:PMC4130781
|
|
|
|
|
35 Kalluri R, Weinberg RA. The basics of
epithelial-mesenchymal transition. J Clin Invest 2009;119(6):1420-1428.
https://doi.org/10.1172/JCI39104
PMid:19487818 PMCid:PMC2689101
|
|
|
|
|
36 Wang L, Wu X, Shi T, Lu L. Epidermal
growth factor (EGF)-induced corneal epithelial wound healing through nuclear
factor κB subtype-regulated CCCTC binding factor (CTCF) activation. J Biol
Chem 2013;288(34):24363-24371.
https://doi.org/10.1074/jbc.M113.458141
PMid:23843455 PMCid:PMC3750138
|
|
|
|
|
37 Kimura H, Okubo N, Chosa N, Kyakumoto S,
Kamo M, Miura H, Ishisaki A. EGF positively regulates the proliferation and migration,
and negatively regulates the myofibroblast differentiation of periodontal
ligament-derived endothelial progenitor cells through MEK/ERK- and
JNK-dependent signals. Cell Physiol Biochem 2013;32(4):899-914.
https://doi.org/10.1159/000354493
PMid:24217646
|
|
|
|
|
38 Huo YN, Chen W, Zheng XX. ROS, MAPK/ERK
and PKC play distinct roles in EGF-stimulated human corneal cell proliferation
and migration. Cell Mol Biol (Noisy-le-grand) 2015;61(7):6-11.
|
|