
・Review
Article・
Molecular
pathobiology of scleritis and its therapeutic implications
Undurti
N Das1,2
1UND Life Sciences, Battle Ground, WA
98604, USA
2BioScience Research Centre and
Department of Medicine, GVP Medical College and Hospital, Visakhapatnam 530048,
India
Correspondence to: Undurti N Das. UND Life Sciences,
2221 NW 5th St, Battle Ground, WA 98604, USA. Undurti@hotmail.com
Received:
Abstract
Scleritis and other
autoimmune diseases are characterized by an imbalance in the levels of
pro-inflammatory and anti-inflammatory molecules with the balance tilted more
towards the former due to the failure of recognition of self. The triggering of
inflammatory process could be ascribed to the presence of cytoplasmic
DNA/chromatin that leads to activation of cytosolic DNA-sensing cGAS-STING
(cyclic GMP-AMP synthase linked to stimulator of interferon genes) pathway and
enhanced expression of NF-κB that results in an increase in the
production of pro-inflammatory bioactive lipids. Bioactive lipids
gamma-linolenic acid (GLA), dihomo-GLA (DGLA), prostaglandin E1 (PGE1),
prostacyclin (PGI2) and lipoxin A4, resolvins, protectins and maresins have
anti-inflammatory actions, bind to DNA to render it non-antigenic and are
decreased in autoimmune diseases. These results suggest that efforts designed
to enhance the production of anti-inflammatory bioactive lipids may form a new
approach to autoimmune diseases. Local injection or infusion of lipoxins,
resolvins, protectins and maresins or their precursors such as arachidonic acid
may be exploited in the prevention and management of autoimmune diseases
including scleritis, uveitis and lupus/rheumatoid arthritis.
KEYWORDS: scleritis;
autoimmune diseases; bioactive lipids; inflammation; micronucleus; cytokines;
resolution of inflammation
DOI:10.18240/ijo.2020.01.23
Citation:
Das UN. Molecular pathobiology of scleritis and its therapeutic implications. Int
J Ophthalmol 2020;13(1):163-175
INTRODUCTION
Scleritis characterized by
inflammation of the sclera, the exterior part of the eye, is usually associated
with auto-immune diseases such as rheumatoid arthritis (RA), lupus, Crohn’s
disease, and other vasculitis. Scleritis is idiopathic and autoimmune
inflammation and infection are the two main causes, though trauma can be an
inciting factor. Despite the fact that scleritis may occur in patients with
autoimmune diseases such as RA, it could be a separate manifestation of the
autoimmune disease. It is well documented that scleritis can sometimes be a
presenting manifestation of a potentially serious systemic disease. At times,
scleritis may precede the systemic disease by many months or even a few years,
one reason as to why it is critical for patients to have regular visits to the
physician/ophthalmologist. Clinical and laboratory evaluation need to be
performed to search for possible autoimmune or infectious causes. Scleral
biopsy and microscopic evaluation can give important information on specific
patterns of inflammation seen and the presence or absence of certain infectious
organisms.
It is not uncommon to have an
extension of scleral inflammation to the anterior uveal tract in severe disease
with ocular complications that may lead to progressive visual loss. In general,
occurrence of anterior uveitis in the course of scleritis indicates poor
prognosis. Hence, the anterior uveal tract should be evaluated at every
follow-up visit of a patient with scleritis, and if, anterior uveitis is noted
it is imperative to start systemic immunotherapy[1].
There are two main types of
scleritis: anterior and posterior scleritis (PS). Anterior scleritis, the most
common type, affects the front part of the sclera and is of three types:
diffuse scleritis, the most common type that causes widespread redness and
inflammation throughout the whole or front portion of the sclera; nodular
scleritis, is known for nodules, often tender to the touch, on the surface of
the eye; necrotizing scleritis, the most severe form of anterior scleritis that
can destroy scleral tissues and may lead to loss of the eye(s) and causes
extreme pain and tenderness (although a rare form can occur without pain). PS,
the rarer form, affects the back part of the eye and often not related to an
autoimmune disease. It can develop on its own or with the anterior form of
scleritis. It is characterized by pain and tenderness and often associated with
complications resulting in retinal detachment and angle-closure glaucoma.
In view of the uncommon nature of
PS, which is often misdiagnosed or under-diagnosed, Dong et al[2], performed a retrospective systemic evaluation of the clinical features, associated
systemic diseases, and risk factors in those with PS with retinal detachment
and evaluated choroidal thickness (CT) noninvasively employing enhanced depth
imaging optical coherence tomography (EDI-OCT) in PS with serous retinal
detachment. Their results revealed that PS with retinal detachment can present
with a variety of symptoms and concluded that typical T-sign detected by B-scan
ultrasound is a useful confirmatory sign for diagnosing PS. It was observed
that pathological increases in CT may be useful as a potential predictor of
inflammation in PS. Despite their thorough clinical and imaging evaluation of
the patients using best corrected visual acuity (BCVA), intraocular pressure
(IOP), fundus examination, posterior coats thickness (PCT) determination by
B-scan ultrasound, and CT measurement by EDI-OCT, no efforts have been made to
evaluate inflammatory markers such as cytokines, and did not report the results
of the treatments offered to these subjects. It would have been helpful had the
authors documented and reported the response of the study subjects to various
treatments offered (such local and systemic steroids, immunosuppressive
therapy, biologics used, etc.) and the corresponding prognosis. In these
days of investigative medicine, it is important that clinicians collaborate
with scientists who have knowledge of molecular biological approaches to
various diseases and identify whether such molecular approaches will give
better clues to the underlying pathobiology of a disease that may lead to a
better understanding of the specific condition and development of novel
approaches to diagnosis and management.
Self and Non-self-discrimination by
Immune System There are two critical issues that
need close scrutiny in scleritis and other eye-related autoimmune diseases
including uveitis: 1) what makes these tissues antigenic; 2) what inflammatory
markers/events occur that can be exploited in their therapy, to predict
prognosis and evaluate response to treatment offered. It will be interesting if
the molecular and biochemical events of the disease could be correlated to the
clinical picture.
One of the cruxes in autoimmune
diseases, in general, and, in specific, eye-related autoimmune diseases such as
scleritis and uveitis is why and how self is recognized as non-self to mount an
immune attack. This suggests that under some very specific circumstances, DNA
(since anti-DNA and DNA-related antibodies are present in majority of the
autoimmune diseases) becomes antigenic.
Our immune system has evolved a
complex mechanism to discriminate between self and non-self. This innate
recognition system is mainly based on receptors that are meant to recognize
non-self-molecules present in pathogens or invading organisms or even tumor
cells but are not present in the host. Thus, innate receptors are meant to
recognize self-molecules that are not present in a healthy state but present in
diseases. This suggests that innate immune system is capable of recognizing
changes that occur in a normal cell when it is infected. The “missing self”
hypothesis proposes that target cells expressing major histocompatibility complex
(MHC) class I molecules are (more) resistant to natural killer (NK) cell
mediated killing compared to virally infected cells which have lost the
expression of MHC class I molecules implying that NK cells are able to
differentiate “self” from “missing self”. In humans, these inhibitory receptors
are represented by immunoglobulin (Ig)-like receptors (KIRs) and lectin-like
CD94/NKG
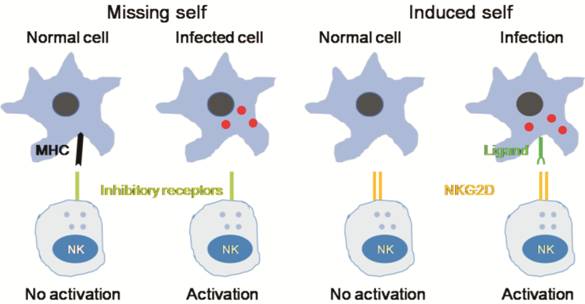
Figure 1 Innate immunity recognizes
changes in cells induced by infection
NK cells
have inhibitory receptors on their surface to differentiate “self” from
“missing-self”. The lack of expression of MHC class I molecules (missing self)
promotes the activation of NK cells and subsequent lysis of the target cells.
NK cells also express activating receptors such as NKG2D that directly
recognize ligands induced in response to infection (induced self). NK
cell-mediated cytotoxicity against malignant target cells or infected cells is
regulated by both activating and inhibitory cell surface immunoreceptors. In
humans, such receptors are of three types: 1) the killer immunoglobulin
receptors (KIRs), 2) natural cytotoxicity receptors (NCRs), and 3) the c-type
lectin receptors. NKG2D is one member of the c-type lectin-activating receptor
family that is evolutionarily conserved and is located within the NK gene
complex on human chromosome 12p12-p13. NKG2D is expressed on all NK cells and
is a promiscuous receptor that recognizes at least 6 counter ligands that
include: the MHC class I-like molecules, MICA and MICB, and members of the ULBP
family (ULPB1-4), named for the ability of some members to bind to the UL-16
protein of cytomegalovirus (CMV).
The adaptive immune response is
different from innate that has the ability to mount a specific immune response
against any microbe or stimuli that our body encounters. The T and B cells that
constitute the adaptative immunity are able to somatically generates large
repertories of specific receptors [(T cell receptor (TCR) and B cell receptor
(BCR)] that have the ability to virtually recognize any non-self-antigen. As a
consequence of this specific and efficient adaptive immune system, it is
essential to discriminate self from non-self in order to avoid
anti-self-reaction (in the form of autoimmune reactions). Thus, lymphocytes
that bear these high avidity autoreactive receptors need to be eliminated or
suppressed or regulated by an efficient negative feed-back control system.
Since such a process is likely to be imperfect, it explains the high frequency
of autoimmune diseases seen in the clinic. At the same time, the adaptive
immune system needs to develop effect or mechanisms that are capable of
eliminating pathogens that calls for it to be coupled with the innate immune
system and use the same system to eliminate the attacking pathogens.
Cytoplasmic DNA and
Inflammation It is evident from the preceding
discussion that autoimmunity is due to failure in self and non-self
discrimination. Several pathogenic mechanisms proposed for the development of
autoimmune diseases include molecular mimicry, exposure of hidden antigens,
loss of suppressor cell function, T and B cell dysfunction, epitope spreading
and epitope drift and polyclonal B cell activation by superantigens. In this
context, it is noteworthy that recognition of microbial nucleic acids by the
host is an important strategy that is needed to respond to various infectious
agents. Several microbial DNAs are introduced into the host cells during infections
that need to be recognised appropriately and eliminated without triggering
abnormal immune responses. The intracellular DNA that is introduced into cells
during infections are known to trigger inflammatory responses by triggering
induction of anti-viral type I interferons (IFNs), tumor necrosis factor-α
(TNF-α), interleukin (IL)-1β and IL-18. If nucleases such as DNase II or DNase
III (Trex1) fail to clear cytosolic DNA, then accumulated DNA drives
inflammatory responses that lead to autoimmune diseases. There appears to exist
various ways to recognize and respond to cytosolic DNA. Thus, it is imperative
that there are specific sensors that can couple cytosolic DNA recognition to
immune signaling. One such pathway leads to the proteolytic activation of the
cysteine protease caspase-1 that is associated with maturation and secretion of
the IL-1β and IL-18. The second pathway could involve the transcriptional
induction of type 1 IFN and pro-inflammatory genes (Figure 2). IL-1β activates
neutrophils, macrophages, dendritic cells, and T cells whereas IL-18 incites
IFN-γ production by NK and T cells. All these events ultimately lead to the
formation of inflammasome that occurs as a result of immune responses to
intracellular DNA of bacterial or viral origin[12-16]. These results suggest that while DNA-induced immune
responses are critical to immunity, failure to recognize self-DNA can lead to
inappropriate consequences namely autoimmune diseases such as lupus in which
type I IFN and autoantibodies directed against dsDNA, RNA and nucleosomes can
be found. Thus, failure of the multiple fail-safe mechanisms employed by the
host are subverted leading to DNA-induced immune responses and inflammation
(Figure 2). One such regulation provided by the body include cellular
endonucleases such as DNase I, DNase II and DNase III (also known as Trex1)
that are normally involved in the clearance of extracellular, lysosomal and
cytosolic DNA respectively. This implies that functional defects in these
enzymes are present in lupus and other autoimmune diseases.
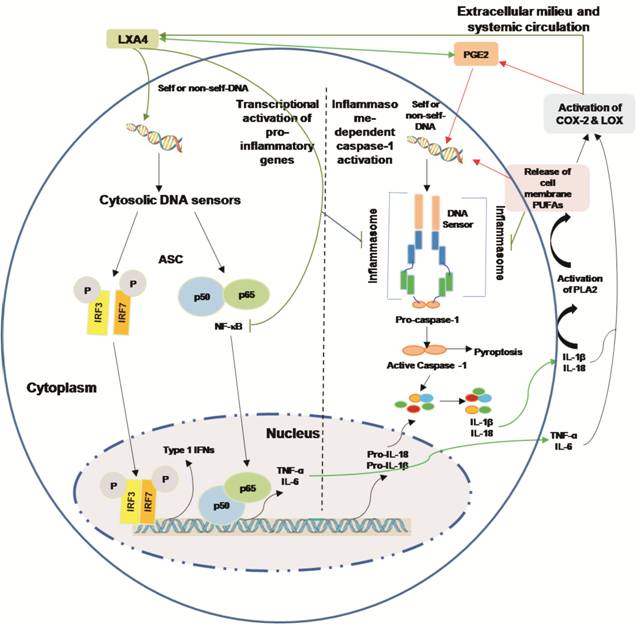
Figure 2 Cytoplasmic DNA triggers
transcription of inflammatory genes and inflammasome-dependent proteolytic
activation of caspase-1 Presence of DNA in the
cytoplasm leads to the activation of two distinct signaling pathways 1)
activation of IRF3, IRF7 and NF-κB that results in the transcriptional
induction of type 1 IFN genes or pro-inflammatory genes IL-6 and TNF-α; 2)
cytosolic DNA leads to the assembly of inflammasome leading to caspase-1
activation and subsequent cleavage of pro-IL-β and pro-IL-18 that results in
the formation of biologically active and mature forms of IL-1β and IL-18.
Caspase activation mediates the cell death under some very specific conditions
(such as tumor cell death). Enhnced synthesis and secretion of pro-inflammatory
cytokines leads to activation of COX-2 and LOX enzmes resulting in the
produciton of pro-inflammatory bioactive lipids such as PGE2, LTs and TXs and a
simultaenous decrease of anti-inflammatory LXa, resolvins, protectins and maresins
from their respetive precursor PUFAs (AA, EPA and DHA). These PUFAs, LXA4,
resolvins, protectins and maresins can suppress NF-κB and IRF3 and IRF7
activation and inhibit the production of pro-inflammatory cytokines. Under some
very speific situations PGE2 may function as an anti-inflammatry molecule. PGE2
and IL-1β have been shown to trigger release of AA/EPA/DHA from the cell
membrane pool by activating PLA2 that can lead to the formation of
anti-inflammatory LXA4/resolvins/protectins/maresins that, in turn, suppress
production of IL-6 and TNF-αto restore homeostasis. Thus, there is a very close
and a positive and negative feed-back regulation between pro- and
anti-inflammatory cytokines, bioactive lipids and respective signaling
pathways.
The fact that cytoplasmic DNA could
incite inflammatory process is supported by the recent studies that showed that
cytoplasmic chromatin (chromatin is a complex of DNA, RNA, and protein found in
eukaryotic cells) activates the innate immunity cytosolic DNA-sensing cGAS-STING
(cyclic GMP-AMP synthase linked to stimulator of interferon genes) pathway,
that leads to two downstream events: type 1 IFN through IRF3, and
pro-inflammatory response through NF-κB (Figure 2). Further studies showed that
cGAS-STING pathway is connected to the NF-κB-mediated senescent associated
secretory phenotype (SASP) that results in the secretion of pro-inflammatory
cytokines (into the surrounding milieu and systemic circulation), recruitment
of immune cells, modulate their activity and consequently alters tissue
microenvironment[17-20]. It is
noteworthy that when wild-type and Sting-null mice are exposed to
sub-lethal ionizing radiation to induce DNA damage, senescence and SASP
program, the production of IL-1α was significantly reduced in the null mice
indicating that STING mediates DNA damage-induced SASP and tissue inflammation[17-18]. Subsequently, it was noted
that STING is essential for Ras-induced SASP and immune surveillance since
expression of STING restored cytokine expression, inflammation and immune
mediated clearance of malignant cells. It is interesting that short-term
inflammation and senescence serve as barriers to tumorigenesis, persistent
inflammation produces tissue damage and enhances tumor growth that explains
increased incidence of cancer (especially lymphomas) in those with lupus,
Behcet’s disease and rheumatoid arthritis (RA) (conditions in which micronuclei
cells are common and are frequently associated with uveitis and scleritis) and
cancer cells frequently contain extra-nuclear chromatin[21-24]. But, paradoxically, cytoplasmic chromatin (DNA)
incidence was never studied in those with scleritis and uveitis despite the
fact that these are autoimmune diseases.
Cytokines and Essential Fatty Acid
Metabolism The cytoplasmic DNA/chromatin
(micronuclei) that is frequent in lupus and RA and other autoimmune diseases
(possibly in uveitis and scleritis)-induced increase in the production of
proinflammatory cytokines IL-1β, TNF-α, IL-18, IL-6 and IFN-γ that spill over
into the intercellular surrounding milieu and systemic circulation can be
detected in the form of their enhanced plasma levels. In view of the putative
role of TNF-α and IL
Essential Fatty Acids
Metabolism Our diet is rich in essential fatty
acids (EFAs) linoleic acid (LA, 18:2 n-6) and alpha-linolenic acid (ALA, 18:3
n-3) and are acted upon by delta-6-desaturase and delta-5-desaturase and
respective elongases to form their long-chain metabolites gamma-linolenic acid
(GLA, 18:3 n-6), dihomo-gamma-linolenic acid (DGLA, 20:3 n-6) and arachidonic
acid (AA, 20:4 n-6) and eicosapentaenoic acid (EPA, 20:5 n-3) and
docosahexaenoic acid (DHA, 22:6 n-3) respectively. LA, GLA, DGLA, AA, ALA, EPA
and DHA are called as polyunsaturated fatty acids (PUFAs) but only LA and ALA
are EFAs since they cannot be formed in the body. DGLA forms the precursor of
prostaglandins (PGs) of 1 series; AA forms the precursor of 2 series PGs,
thromboxanes (TXs) and 4 series leukotrienes (LTs), whereas EPA is the
precursor of 3 series PGs, TXs and 5 series LTs. Most of the PGs, TXs and LTs
are pro-inflammatory in nature (2 series PGs and TXs>3 series PGs and TXs
and 4 series LTs>5 series LTs). The conversion of DGLA, AA and EPA to their
respective PGs, TXs and LTs is due to the action of cyclo-oxygenases (COX-1 and
COX-2) and 5-, 12- and 15-lipoxygenases (5-LOX, 12-LOX and 15-LOX). It is
noteworthy that AA is also the precursor of a potent anti-inflammatory product
lipoxin A4 (LXA4) while EPA is the precursor of similar anti-inflammatory
metabolites called as resolvins whereas DHA gives rise to resolvins, protectins
and maresins[34] (Figure 3).
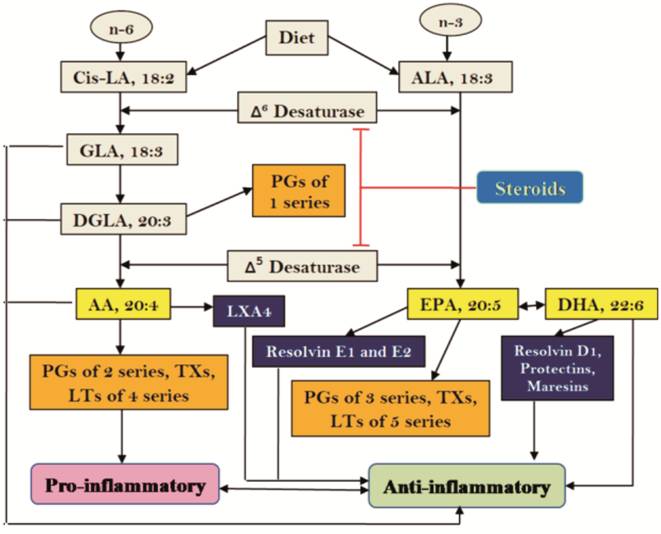
Figure 3 Scheme showing metabolism
of essential fatty acids and their pro- and anti-inflammatory products Steroids block the activities of
desaturases that leads to a decrease in the concentrations of GLA, DGLA, AA,
EPA and DHA. This results in decreased formation of not only pro-inflammatory
PGs, LTs and TXs but also lipoxins, resolvins, protectins and maresins (due to
precursor deficiency). Hence, resolution of inflammation will be incomplete.
This ultimately causes continuous of inflammation to chronic phase and failure
of healing of wound due to deficiency of lipoxins, resolvins, protectins and
maresins.
Interaction(s) among cytokines,
phospholipases, PUFAs, COX-2, LOX enzymes, corticosteroids and their relevance
to inflammation and resolution of inflammation It is likely that under normal
physiological conditions, a delicate balance is maintained between pro- and
anti-inflammatory eicosanoids (similar to the balance struck between pro- and
anti-inflammatory cytokines). It is interesting to note that when the
inflammatory process reaches its peak, the anti-inflammatory pathway is
triggered that results in the formation of adequate amounts of LXA4, resolvins,
protectins and maresins and anti-inflammatory cytokines accompanied by
suppression of reactive oxygen species (ROS) generation and enhancement of
anti-oxidant defences that results in the resolution of inflammation and
restoration of homeostasis. Lipoxins, resolvins, protectins and maresins are
essential for resolution of inflammation and wound healing since they inhibit
polymorphonuclear leukocytes (PMNLs) trans-endothelial migration, reduce
leucocyte infiltration, and suppress dendritic cells (DC) migration and IL-12
production. Lipoxins, resolvins, protectins and maresins enhance the expression
of anti-inflammatory genes and attenuate LTB4-stimulated proinflammatory
signals[34]. In general, lipoxins, resolvins,
protectins and maresins seem to have similar and overlapping anti-inflammatory
and pro-resolving actions.
In this context, the interactions
between pro- and anti-inflammatory cytokines and PUFAs metabolism is
interesting. Proinflammatory cytokines IL-1, IL-6, TNF-α and IFN-γ activate
phospholipases, enhance ROS generation[35-39],
increase activity of COX-2 and LOX enzymes to augment the production of
pro-inflammatory PGE2, TXA2 and LTs. The precursors for the formation of these
proinflammatory eicosanoids are derived from the cell membrane lipid pool by
the activation of phospholipase A2 (PLA2) by pro-inflammatory cytokines
(Figures 2 and 3). It is important to note that the release of PUFAs from the
cell membrane lipid pool occurs in two waves by their respective
phospholipases. There are three classes of phospholipases that regulate the
release of PUFAs: calcium-independent PLA2 (iPLA2), secretory PLA2 (sPLA2), and
cytosolic PLA2 (cPLA2). Each class of PLA2 is further divided into isoenzymes
for which there are 10 for mammalian sPLA2, at least 3 for cPLA2, and 2 for
iPLA2. The first wave of release of PUFAs from the cell membrane occurs due to
the action of iPL2 that leads to the formation of pro-inflammatory PGE2, TXA2
and LTB4. The second wave of release of PUFAs occurs by the action of sPLA2 and
cPLA2 at the time of resolution of inflammation leading to the formation of
lipoxins, resolvins, protectins and maresins that are essential for the
suppression of inflammation. It is paradoxical to know that formation of
adequate amounts of PGE2 is necessary to both induce optimal inflammation and
at the same time to trigger the initiation of resolution of inflammation. Thus,
PUFAs released at the instance of inflammatory stimuli by the activation of
iPLA2 are directed to form pro-inflammatory PGs, TXs and LTs whereas PUFAs
released from the cell membrane at the time of resolution of inflammation
triggered by sPLA2 and cPLA2 are directed to form lipoxins, resolvins,
protectins and maresins. This delicate balance and switch over from
pro-inflammatory to anti-inflammatory molecules are determined by the type of
PLAs that are activated in response to pro- and anti-inflammatory cytokines and
the activities of COX-2 and 5-, 12- and 15-LOX enzymes. This close co-operation
and interaction(s) among PLAs, COX-2, LOX enzymes and various cytokines is
essential for the appropriate inflammation to occur for gradual, smooth and
orderly onset of anti-inflammatory events and resolution of inflammation and
restoration of homeostasis[34]. Any defects in
this process (dysfunction of cytokines, PLAs, COX, LOX enzymes, cell membrane
stores of PUFAs, etc.) could result in persistance of inflammation and
damage to the target tissues as is seen in autoimmune diseases including
scleritis and uveitis.
In this context, it is important to
note that IL-6, TNF-α and corticosteroids suppress the activities of
desaturases resulting in deficiency of AA, EPA and DHA that results in
decreased formation of LXA4, resolvins, protectins and maresins (due to
precursor deficiency) but ironically excess formation of PGs, LTs and TXs
persists[34]. In contrast, IL-6 and TNF-α
activate PLA2, COX-2 and LOX enzymes whereas corticosteroids suppress them.
This is supported by the observation that supplementation of AA during active
inflammatory process when PGs, LTs and TXs are being synthesized in excess
actually results in an increase in the formation of LXA4 (and possibly,
resolvins, protectins and maresins) with or without any change in the
concentrations of PGE2 (and thus, tilting the balance more towards
anti-inflammatory molecules) and suppresses the inflammation process[34,40-41]. AA, EPA,
DHA, LXA4, resolvins, protectins and maresins inhibit the production of IL-6,
TNF-α and ROS. Thus, corticosteroids, IL-6 and TNF-α can induce an EFA
(PUFAs)-deficiency state by their ability to block the activities of
desaturases as a result formation of lipoxins, resolvins, protectins and
maresins is decreased leading to failure of resolution of inflammation. In the
initial stages of inflammation, corticosteroids suppress inflammation by
blocking the activities of PLA2, desaturases, COX-2 and LOX enzymes. In
contrast, IL-6 and TNF-α induce inflammation by activating PLA2, COX-2 and LOX
enzymes and enhancing the formations of PGs and LTs. This may explain why
steroids are potent suppressors of acute inflammation but in the long run fail
to induce wound healing since they block the formation of LXA4, resolvins,
protectins and maresins that are needed for resolution of inflammation[34,42-43]. IL-1β
that is markedly increased during the inflammatory process induces PG
biosynthesis and also up regulates the formation of LXA4 and maresins that are
needed for resolution of inflammation. Both LXA4 and maresins (resolvins and
protectins) are potent down-regulators of PGE2 production. Increased
15-prostaglandin dehydrogenase (15-PGDH) expression seems to augment the
formation of LXA4, resolvins, protectins and maresins and regeneration of
tissues to aid reestablish tissue homeostasis[34,42-48]. Thus, IL-1β and PGE2 seem to
have both pro- and anti-inflammatory actions depending on the context (Figure
4). These results imply that in order to suppress acute and chronic
inflammation and inhibit the production of pro-inflammatory IL-6 and TNF-α, one
need to employ LXA4, resolvins, protectins and maresins in combination with
corticosteroids with/without AA/EPA/DHA in inflammatory conditions such as
scleritis and uveitis.
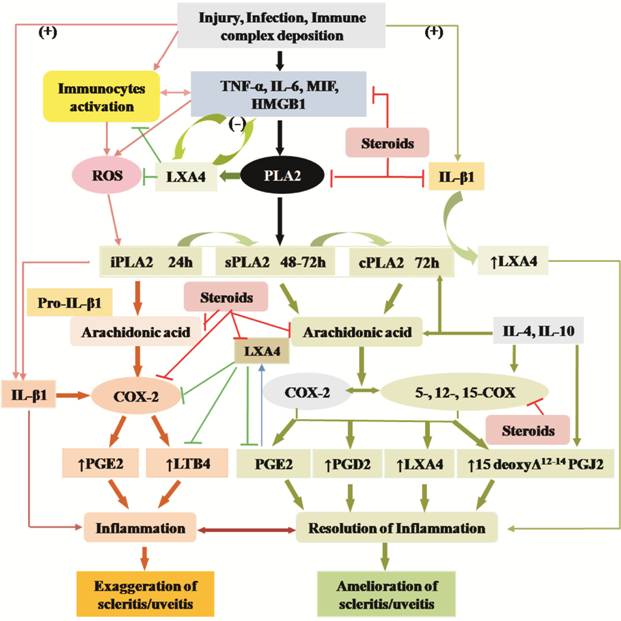
Figure 4
Scheme showing the relationship among pro- and anti-inflammatory cytokines,
PGs, LTs, lipoxins, resolvins, protectins and maresins and steroids. Metabolism
and actions of arachidonic acid is shown as a representative of various PUFAs
(DGLA, EPA and DHA) (+) Indicates increase in the
synthesis/action or positive effect. (-) Indicates decrease in the
synthesis/action or negative effect.
PUFAs and their metabolites regulate
cytoskeleton system Since PUFAs (especially GLA, DGLA,
AA, EPA and DHA) and their pro- and anti-inflammatory metabolites are potent
regulators of the expression and concentrations of pro- and anti-inflammatory
cytokines, ROS, and pro- and anti-inflammatory events, it is important to know
mechanism(s) of their action. In addition to their ability to enhance or
suppress the activation of various immunocytes (PMNLs, T cells, macrophages,
dendritic cells, etc.), it is noteworthy that these bioactive lipids can
alter the cell membrane (not only cell membrane, but also that of mitochondrial
membrane, nuclear membrane, etc.) fluidity by virtue of their
incorporation into it. Thus, when the membrane content of PUFAs is high it will
become more fluid and when their PUFAs content is low (this is likely to happen
when saturated fatty acids and cholesterol content of the membrane is increased
at the cost of PUFAs) the membrane will be more rigid. It was reported that
increasing the PUFAs content of the cell membranes not only increases membrane
fluidity but also increases the number of insulin receptors and their affinity
to its receptors, whereas higher content of saturated fatty acids decreases
membrane fluidity and decreases the number of insulin receptors and their
affinity to the receptors[49-54].
These studies imply that PUFAs can alter cell membrane fluidity and thus, alter
the expression of receptors and their binding to their respective receptors.
Cell membrane configuration/properties are controlled by elements of the
cytoskeleton (that include microfilaments, microtubules, and intermediate
filaments) that play a critical role in maintaining the cell shape, cell
movement, intracellular transport/trafficking, cytokinesis and cytoplasmic streaming.
Since PUFAs and their metabolites regulate phagocytosis, cell mobility, cell
receptor number and secretion of cytokines and other molecules, it is
reasonable to propose that these bioactive lipids can modulate cytoskeleton
system. It was reported that treatment of mouse lymphocytes with LA produced
alterations in their cytoskeleton and contractile proteins indicating that
PUFAs have the ability to alter the interaction of surface receptors with the cytoskeleton
and thus, could affect cytoplasmic distribution of the proteins[55-56]. It is interesting to note that
15 deoxyΔ12-14 PGJ2 that is essential for resolution of inflammation
and has potent anti-inflammatory actions with proteins that are involved in
cytoskeletal organization, such as actin, tubulin, vimentin, and tropomyosin
and induced early reorganization of vimentin and tubulin in cultured mesangial
cells of the kidney[57].
12(S)-hydroxyeicosatetraenoic acid (12-HETE), derived from AA, that has
chemotactic actions on neutrophils and macrophages is known to influence
glucose transporter 4 (GLUT4) translocation and thus, regulate glucose
transport by contributing to rearrangement of actin cytoskeletal elements[58]. HETE is important for corneal epithelial cell
migration during wound healing[59] suggesting
that AA metabolites participate in the restoration of functional integrity of
cornea and sclera once the inflammation process subsides. It is noteworthy that
LXA4, a potent anti-inflammatory metabolite of AA that is known to modulate
leukocyte trafficking and stimulate nonphlogistic macrophage phagocytosis of
apoptotic neutrophils and thus, promotes the resolution of inflammation and
wound healing has been shown to facilitate actin cytoskeleton rearrangement and
cell polarization[60]. These evidences[55-60] highlight the critical role of
PUFAs and their metabolites not only in inflammation and its resolution,
stimulation of nonphlogistic macrophage phagocytosis of apoptotic neutrophils
and debris removal at the site of inflammation, but also their regulatory role
incorneal and scleral cell proliferation, migration and restoring their
(corneal and scleral) functional integrity.
PUFAs and their metabolites and
micronuclei cells Cytoplasmic DNA/chromatin triggers
inflammationby enhancing the expression of NF-κB and such micronuclei cells are
frequent in autoimmune diseases[16-24]
(Figure 2). In this context, it is noteworthy that studies showed that GLA,
DGLA, PGE1 and PGI2, which have anti-inflammatory actions[61-70] can prevent/decrease radiation, benzo(a)pyrene, and
other chemicals-induced incidence of micronucleus containing human lymphocytes
and mouse bone marrow cells[71-78].
These results suggest that PUFAs and their anti-inflammatory metabolites
including LXA4 can prevent the generation of cytoplasmic DNA/chromatin in cells
and thus, prevent inflammation and consequent autoimmune process. In addition,
PUFAs and their metabolites seem to be capable of binding to DNA and regulate
the expression of several genes to bring about their critical actions[79-82]. Our recent studies revealed
that PUFAs and their metabolites especially LXA4 can suppress the expression of
NF-κB and alter the expressions of p53, Ras, Myc, Ros, Ras,
Bax, Bcl-2, caspases, lipocalin-2, PDX1, Nrf2,
GLUT-2 and other genes in vitro and in vivo[83-87]. It was noted that peroxidized
products of PUFAs but not PUFAs bind to DNA and thus, regulate gene(s)
expression[88-90]. These
results suggest that PUFAs and their metabolites such as PGs and LXA4 bind to
DNA not only to regulate gene expression but also to render DNA non-antigenic
to prevent cytoplasmic DNA/chromatin-induced inflammatory process. Thus, PUFAs
and their metabolites may aid T cells and other immunocytes in self and
non-self-discrimination. This may explain the apparently paradoxical results
obtained by us wherein GLA supplementation to patients who are on long-term
treatment with DPH (diphenylhydantoin for epilepsy) showed decreased number of
micronuclei containing peripheral lymphocytes but DNA ladder pattern (an
indication of DNA damage) was increased suggesting apoptosis of cells harboring
DNA damage. This implies that GLA prevents cells from accumulating genetic
damage[77].
CONCLUSIONS AND THERAPEUTIC IMPLICATIONS
It is evident from the preceding
discussion that cytoplasmic DNA/chromatin triggers pro-inflammatory process by
enhancing the expression of NF-κB that, in turn, leads to enhanced production
and secretion of IL-1β, TNF-α, IFNs, IL-10 and IL-6 (Figure 2), which
ultimately results in the onset of autoimmune diseases lupus, RA, scleritis and
uveitis[16-24]. It is known
that in auto-immune diseases the number of buccal mucosal cells and circulating
lymphocytes containing
micronuclei is increased[21-24]. The enhanced secretion of
pro-inflammatory cytokines upregulates the activities of PLA2, COX-2 and LOX
enzymes that leads to an increase in the production of pro-inflammatory PGs,
LTs and TXs and decrease in anti-inflammatory bioactive lipids lipoxins,
resolvins, protectins and maresins. Thus, there is a cross-talk between
cytokines and bioactive lipids in the pathobiology of auto-immune diseases
including scleritis and uveitis. Hitherto, it has been customary to measure
plasma, synovial and tissue fluid(s) content of cytokines, PGs, LTs and TXs to
assess and quantify the inflammatory process. Based on the preceding
discussion, I propose that the number of micronucleus containing cells, plasma
and tissue fluid(s) content of PGs, LTs, TXs, lipoxins, resolvins, protectins
and maresins in addition to the concentrations of cytokines could be employed
to assess the degree of inflammation, response to therapy and prognosis of the
underlying auto-immune disease(s). Thus, it is recommended that in those with
scleritis (and other auto-immune diseases): 1) Measure the number of
micronucleus containing cells in the scleral scrapings and other appropriate
tissues. 2) Estimate the concentrations of various cytokines and bioactive
lipids (PUFAs, PGs, LTs, TXs, lipoxins, resolvins, protectins and maresins) in
the tears, plasma and other body fluids (such as vitreal fluid in the case of
uveitis). 3) These measurements could be used to assess the degree of
inflammation, and changes in their concentrations may aid in assessing the
progress of the disease, response to therapy and prognosis. It is likely that
if the number of micronucleus containing cells and pro-inflammatory cytokines
and PGs, LTs, TXs are decreasing with therapy the patient is responding to the
treatment offered and prognosis is good. Furthermore, the number of micronucleus
containing cells could be correlated with the cytokine profile and
concentrations of bioactive lipids to assess the balance between pro- and
anti-inflammatory events to assess response to therapy, progress and prognosis
of the disease.
Measuring the concentrations of
various cytokines and bioactive lipids in the tears of patients with scleritis
and other inflammatory conditions of eye (such as uveitis wherein vitreal fluid
can be used for such measurements) will form a simple, elegant and objective
method of both clinical and laboratory assessment of progress of the disease
that could be correlated with the clinical picture. Wherever facilities and
expertise permit, perhaps, cellular scrapings (or cell/tissue samples) could be
used for measuring the expressions of NF-κB, various cytokines, desaturases,
COX and LOX enzymes, PG synthetases and other gene expression studies and
correlated with the concentrations of their products and correlated to the
clinical picture. Such in depth studies employing molecular, biochemical and
genetic studies would enhance our understanding of the pathogenesis of the
diseases.
In addition, the preceding
discussion implies that newer therapeutic approaches in the management of
scleritis and uveitis and other ophthalmic inflammatory conditions is a
distinct possibility. LXA4/resolvins/protectins/maresins containing ophthalmic
preparations could be developed and further studies could investigate the
potential role of their local instillation (on the surface of the eye or
intravitreally, especially for those with uveitis and possibly, diabetic
retinopathy as it is also considered as an inflammatory condition[91-93]) in reduction of inflammation
by themselves alone or in combination with local and systemic steroids and/or
with immunosuppressive drugs to induce or enhance the anti-inflammatory process
and accelerate healing. Such novel therapeutic approaches could be attempted in
future.
ACKNOWLEDGEMENTS
Conflicts of Interest: Das UN, None.
REFERENCES